New variant of PRDM16 gene nucleotide sequence in a family with various phenotypic manifestations of the non-compacted myocardium
Left ventricular noncompaction (LVN) is a rare disorder characterized by an abnormal myocardial structure with a pronounced noncompact layer and increased trabecularity [1]. In recent years, the incidence of LVN has increased significantly due to improvement of imaging methods. However, along with this, the complexity of correct clinical interpretation of this phenomenon is also growing. There are different conditions with LVN, which varies from primary cardiomyopathies (CMP) and other congenital diseases leading to severe heart failure (HF) and requiring radical treatment, to incidental finding in people without complaints (for example, athletes and pregnant women) [2]. In this regard, the assessment of LVN morbidity, the determination of its contribution in decompensated HF and the prognosis in each case are possible only after a comprehensive analysis of the clinical performance, family and genetic history [3][4].
According to current estimates, at least half of LVN cases are hereditary; in about 30% of cases, it is possible to detect a pathogenic or potentially pathogenic genetic variant by DNA diagnostics [1]. Genetic determinism, i.e. the presence of genetic variants mediating the LVN is classified as an independent risk factor for fatal cardiovascular events and an unfavorable prognosis of CMP [2]. In this regard, genetic testing is recommended for patients with an established diagnosis of LVN [5]. To date, at least 80 genes are associated with LVN, and this list is expanding with the accumulation of next-generation sequencing (NGS) data. Although a significant proportion of the variants detected in patients with LVN is attributed to cardiac sarcomere protein genes, which are also associated with various types of primary CMP (in particular, MYH7, MYBPC3, TTN). The variety of genetic findings in LVN is much wider and includes a large number of variants in genes encoding many factors of cell development and differentiation, including factors of myocardial embryogenesis, etc. These data support the hypothesis of LVN embryonic origin, however, to clarify the role of such genes and increase the evidence of their relationship with LVN, more data on genotype-phenotype correlations are needed.
This article presents a family with different phenotypic manifestations of LVN with the same polymorphism encoding the transcription factor responsible for after-birth suppressing the expression of genes involved in prenatal development (Figure 1).

Figure 1. Pedigree.
Case report
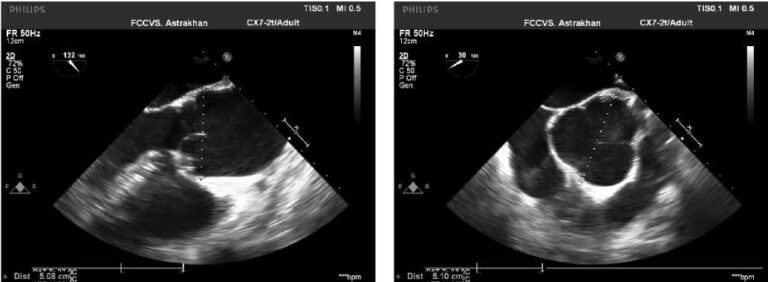
The proband is a 33-year-old normosthenic woman who is being followed up by a cardiologist at the National Medical Research Center for Therapy and Preventive Medicine. At the age of 27, she began to feel palpitations during the first pregnancy. Then, there was a short of breath, dizziness. In October 2015, she underwent examination at the National Medical Research Center for Therapy and Preventive Medicine. In blood tests, all the standard parameters were within the normal range. Twenty-four hour Holter monitoring (24HM) revealed rare premature ventricular contractions (PVCs). Echocardiography revealed end diastolic dimension (EDD) of 5,2 cm, interventricular septum thickness (IVST) of 0,8 cm, left ventricle (LV) ejection fraction (LVEF) of 47%, as well as the signs of LVN in the apex and lateral wall (Chin, Stollberger, Jenni criteria). Contrast-enhanced cardiac magnetic resonance imaging (MRI) (Figure 2) showed data suggestive of a myocardial noncompaction without cavity dilatation (LVEF 45%). Betablockers, angiotensin-converting enzyme inhibitors, and mineralocorticoid receptor antagonists were prescribed. After hospitalization, the patient felt satisfactory and did not take medications all the time. There was deterioration since the spring of 2017, when the palpitations and weakness appeared. According to 24HM with bisoprolol therapy (2,5 mg a day), a sinus rhythm with a heart rate (HR) of 46-78-144 bpm, 6640 isolated PVCs, 10638 bigeminy episodes, 40 coupled PVCs were detected, no pauses were recorded. Echocardiography revealed EDD of 5,3 cm, IVST of 0,8 cm, LVEF of 38%, type 2 diastolic dysfunction, signs of LVN in the apex and lateral wall (Chin, Stollberger, Jenni criteria) (Chin, Jenni, Stollberger criteria [6-8]). The therapy was altered as follows: increasing bisoprolol dose, adding spironolactone and perindopril. The patient’s condition stabilized. With annual dynamic follow-up, according to echocardiography, the normal cardiac dimensions and LVEF of 45% are preserved.

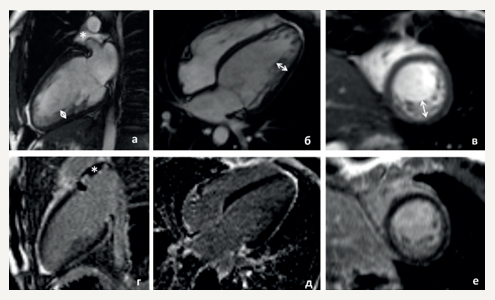
Figure 2. Cardiac MRI of the proband (II-2). (a-c) cine-mode, SSFP sequence: a — long axis 2 chamber view, b — long axis 4-chamber projection, c — short axis, (d-f) — delayed contrast enhancement, IR sequence with suppression of myocardial signal. There are no areas of intramyocardial fibrosis, scarring and post-inflammatory lesions.
Notes: arrows indicate an increase in LV trabecularity in the medial lateral and lower segments; the non-compact and compact layer thickness is 12-15 and 5 mm, respectively; * — a small amount of free fluid in the superior pericardial recess.
The diagnosis of LVN in the proband was established by echocardiography criteria for myocardial noncompaction [9] and was confirmed by MRI criteria (Jacquier and Petersen) [10][11].
Phenotypic cascade screening
Pedigree and clinical data on the proband’s relatives are presented in Figure 1 and Table 1.
Table 1
Clinical data

Abbreviations: CHD — congenital heart disease, MRI — magnetic resonance imaging, left ventricular LVN — non-compaction, TEE — thromboembolic events, EF — ejection fraction, HF — heart failure.
The 38-year-old biological brother of the proband underwent cardiac screening, which did not reveal LVN.
The proband’s maternal normosthenic (height 160 cm, weight 60 kg) half-brother, 25 years old, underwent a comprehensive cardiac examination. All standard blood indicators were within the normal range. Echocardiography revealed EDD of 4,9 cm, IVST of 1,0 cm, LVEF of 53%, as well as signs of myocardial noncompaction in the apex, lateral and posterior walls (Stollberger criterion). According to cardiac MRI (Figure 3), the cardiac cavities were not dilated, the LV contractility was not reduced; there were no fibrosis, scarring and post-inflammatory lesion areas; the myocardial structure was normal, the trabecularity of middle and apical segments of the lateral and posterior LV walls was slightly increased.

Figure 3. Cardiac MRI of the proband’s half-brother (II-4). (a-c) cine-mode, SSFP sequence: a — long axis 2 chamber view, b — long axis 4-chamber projection, c — short axis, (d-f) — delayed contrast enhancement, IR sequence with suppression of myocardial signal. There are no areas of intramyocardial fibrosis, scarring and post-inflammatory lesions.
Note: * — increased LV apex trabecularity due to the “loose” papillary muscle structure.
The 58-year-old proband’s mother. During the third birth, an increase in blood pressure (BP) up to 160/100 mm Hg was noted. She was not further examined and BP was not measured. From the age of 50 years, there was BP increase up to 160-180/100-110 mm Hg. She did not take antihypertensive drugs. For the first time, she was examined at the National Medical Research Center for Therapy and Preventive Medicine at the age of 55. Echocardiography revealed signs of myocardial noncompaction, IVST of 1,2 cm, outflow tract 1,4 cm, LV posterior wall of 1,5 cm, EDD of 5,4 cm, inceased LV trabecularity, especially in the posterolateral LV wall area. The LV myocardium had sponge-like network structure with intertrabecular lacunae stained by Doppler color flow mapping. The ratio of compact (6 mm) and non-compact (18) myocardial layers in the posterior wall area was 3,0 (pronounced myocardial noncompaction according to the Chin, Stollberger, Jenni criteria). Electrocardiography revealed sinus rhythm with a heart rate of 65 bpm, left axis deviation, complete left bundle branch block. Contrast-enhanced cardiac MRI (Figure 4) revealed asymmetric LV hypertrophy (thickness of the basal anterior and anterior septal segments — 13-14 mm; thickness of other basal and middle segments — ?7-9 mm; apical — 4-6 mm), papillary muscle hypertrophy (9-12 mm) with increased trabecularity in the apex and along the anterolateral LV wall with a non-compact and compact layer of 10-20 mm and 4-7 mm thick, respectively. The non-compacted myocardial mass was 16% of compacted one. After the examination, angiotensin-converting enzyme inhibitors and betablockers were prescribed, which the patient takes irregularly. Currently, the BP is 160-170/90 mm Hg.

Figure 4. Cardiac MRI of the proband’s mother (I-4). (a-c) cine-mode, SSFP sequence: a — long axis 2 chamber view, b — long axis 4-chamber projection, c — short axis, (d-f) — delayed contrast enhancement, IR sequence with suppression of myocardial signal. There are no areas of intramyocardial fibrosis, scarring and post-inflammatory lesions.
Notes: arrows indicate a hypertrophied anterior septal segment (14 mm thick); * — arrows indicate an increase in LV trabecularity in the medial lateral segments; the non-compact and compact layer thickness is 15 and 6 mm, respectively; ? — a small amount of fluid in the pe ricardial cavity from the right side.
The proband’s father died suddenly at the age of 35 (prior alcohol abuse).
The 4-year-old proband’s daughter. The girl did not gain weight from 3 months old, then shortness of breath, tachypnea appeared. During a routine exami nation, a congenital heart defect was suspected. Echocardiography revealed LVEF of 35%, increased LV trabecularity, left to right shunt in the central part of interatrial septum measuring 3 mm. Renal ultrasound showed an increase in both kidneys. Therapy was prescribed with prednisolone 5 mg a day, spironolactone 12,5 mg a day, and digoxin 0,05 mg a day. There was no positive response to treatment. To verify the diagnosis, she was referred to the National Medical Research Center for Children’s Health. Echocardiography demonstrated pronounced left heart dilatation (EDD, 3,8; ESD, 3,0 cm), LVEF of 36%, LVN, patent foramen ovale of 1,5 mm. The 24HM revealed sinus rhythm with a heart rate of 80-122-172 bpm, preexcitation phenomeno, an isolated PVC. With selected therapy, the condition improved. She constantly takes amiodarone 50 mg a day, conventional diacarb therapy, digoxin 0,02 mg a day, captopril 2,5 mg 3 times a day, furosemide 3 mg a day, carvedilol 0,78 mg 2 times a day. At present, the myocardial noncompaction manifistations are practically arrested. The child develops normally.
Tables 2 and 3 combine the imaging results of the proband and her relatives.
Table 2
The severity of myocardial non-compaction in the proband and his relatives, according to cardiac MRI

Table 3
Cardiac MRI parameters in the proband and his relatives

Abbreviations: EDV — end diastolic volume, NM — noncompacted myocardium, LVEF — left ventricular ejection fraction.
Genetic cascade screening
Proband and all first-degree relatives underwent molecular genetic analysis.
Whole genome sequencing and bioinformatics analysis
DNA was isolated from whole blood samples using the QIAamp DNA Blood Mini Kit (Qiagen, Germany). The whole genome sequencing (WGS) library was prepared using the Nextera DNA Flex kit (Illumina, USA) according to the manufacturer’s instructions. The average sequencing depth (150 bp) was 30X or more. The reads were aligned to the reference genome (GRCh38), and small variants were found using the Dragen Bio-IT platform (Illumina, USA) and refined using GLnexus [12]. The annotation was carried out using Ensembl VEP [13]. For clinical interpretation, nucleotide sequence variants in LVN-related genes according to the available literature, with frequencies <0,5% in the gnomAD database, were selected. The assessment of the pathogenicity of the variants was carried out in accordance with the current national guidelines for NGS interpretation [14]. Findings were verified by bidirectional Sanger sequencing.
Results of molecular genetic analysis
A molecular genetic analysis revealed a previously undescribed single nucleotide deletion in the PRDM16 gene, leading to a reading frame shift in exon 9 and the formation of a premature stop codon (NM_022114.4: c.2436delT; NP_071397.3: p.Ala813ProfsTer58) (Figure 5). The variant was confirmed in the proband, her daughter, mother and maternal half-brother. Based on the actual pathogenicity criteria, the find was classified as a pathogenic variant (class V). This variant was not found in the proband’s biological brother.

Figure 5. Exon structure of the PRDM16 gene. The correct reading sequence is marked in green, the reading area in the wrong reading frame is marked in blue, and the unreadable area is marked in gray. A yellow marker indicates a mutation, an asterisk indicates a premature stop codon.
Discussion
This paper presents a family with different phenotypes of LVN. The most severe course of the disease is observed in the proband’s daughter, who was diagnosed with dilated LVN with severe heart failure. The proband had an isolated LVN with a slight decrease in LV systolic function to 46%. The clinical picture is dominated by symptomatic cardiac arrhythmias in the form of frequent PVCs, without ventricular tachycardia runs. In the proband’s mother, attention is drawn to myocardial hypertrophy against the background of LVN, which, in turn, may be due to an increases BP. Given the asymptomatic course, the actual onset of hypertension remains unknown, and therefore it is not possible to rule out hypertension as a cause of myocardial hypertrophy against the background of an initially compromised myocardium.
All family members with the LVN phenotype (the proband, her daughter, mother, and maternal halfbrother) were found to have the PRDM16 pathogenic variant, leading to the loss of a gene copy. PRDM16 encodes a transcription factor protein containing zinc finger domains. PRDM16 forms complexes with various transcriptional cofactors and chromatin modulators. Depending on the biological task, it can stimulate or suppress tissue-specific gene expression. The role of PRDM16 has been extensively studied in adipose tissue [15]. Simultaneously, the expression of PRDM16 in mouse and human cardiomyocytes was shown [16][17].
The association of the PRDM16 gene with LVN was first suggested in the study by Arndt A-K, et al. in 2013 [16] based on high prevalence of dilated CMP and LVN in carriers of 1p36 deletion of on chromosome 1, which includes this gene. Subsequent studies have confirmed the role of variants leading to the loss of PRDM16 copy in the development of LVN and dilated CMP in children [18]. In experiments on PRDM16 knockout mice, the role of PRDM16 in the development of hypertrophic CMP was shown [17][19]. Normally, PRDM16 plays a protective role, but the absence of PRDM16, according to Cibi et al. (2020), leads to myocardial hypertrophy, excessive ventricular fibrosis, mitochondrial dysfunction and metabolic disorders in the cell, which contributed to heart failure in adulthood [17]. The effect is mediated by the reactivation of the genetic program of embryogenesis in adult mice, i.e. preservation of high activity of genes involved in embryonic development, but in a deactivated state after birth. In PRDM16 knockout mice, activation of hypertrophic genes NPPA, NPPB, MYH7, MYL14, increased expression of genes involved in fibrosis development (TGFb2, CTGF, TIMP4, LTBP2), genes involved in carbohydrate metabolism (BDH1, PDK4, GLUT1, HMGCS2, PPARG), decreased expression of genes involved in fatty acid oxidation (FASN, CD36, SCD1, SCD2, ADIPOQ), genes responsible for mitochondrial function (mt-ND4, GPAM, UCP3, DLAT, MTHFD2), iron metabolism (TFRC, HAMP, ALAS1, ALAS2, LCN2) and others [17]. Cibi DM, et al. (2020) also showed that hypertrophic CMP could develop in young mice, but in response to metabolic stress [17].
However, there are still too few clinically significant findings for variants in the PRDM16 gene to confidently assess its contribution to disease etiology. In the Clinical Genome Resource database (www.clinicalgenome.org), the level of evidence for a gene-disease relationship for PRDM16 is currently designated as limited. The importance of the PRDM16 gene product for the normal development of cardiomyocytes has been shown in animal models [16]. However, the available data are insufficient for an unambiguous conclusion about the association of this gene with LVN and primary CMP.
Cosegregation of a variant with symptoms within the family is one of the key arguments in favor of the association of this variant with disease and the basis for increasing the pathogenicity class. We believe that our finding confirms the available data on PRDM16 role in the pathogenesis of LVN and indicates the rationale of including this gene in genetic panels for CMP diagnosis. However, the question of the diversity of clinical manifestations within the same family remains open. First, according to the literature, the PRDM16 variants themselves can be associated with both diastolic and hypertrophic CMP (in an animal experiment). There are no explanations for this fact in the literature. Secondly, the severe phenotype in the proband’s daughter allows to expect the presence of a second pathogenic variant, which aggravates the CMP course, but no other potentially significant variants were identified in the child. Thirdly, the early and more severe course of LVN in a proband’s daughter may be the result of metabolic disorders in the neonatal period. There are no clear anamnestic data in favor of this assumption, however, the clinical stabilization and the normal child development over the past three years speak in favor of prior stress factor, which led to metabolic disorders and triggered an episode of significant activation of genes responsible for embryonic development, which was not prevented due to absence of PRDM16 expression.
Conclusion
This paper presents a family with different phenotypic manifestations of LVN in the presence of the same previously undescribed PRDM16 gene variant encoding a transcription factor responsible for afterbirth suppressing the expression of genes involved in prenatal development. The segregation of the symptomatic variant in three family generations testifies in favor of the association of the identified variant with LVN. At the moment, due to the low awareness of PRDM16 gene contribution in the disease development, the influence of its variants on the clinical course and, moreover, about the therapy personalization for carriers of such variants, we cannot in any way change the tactics of patient management based on our findings. However, in the context of the increasingly widespread introduction of highthroughput molecular genetics methods into the patient monitoring strategies with primary heart diseases, the information of such findings, which do not carry any obvious practical benefit at this stage, seems necessary to draw attention to insufficiently studied candidate genes for LVN. The current scheme of DNA diagnostics of CMP often involves focusing attention on well-studied genes. The variant discovered by us and its cosegregation with LVN signs in three generations, together with the previous literature data on PRDM16 findings, indicate that this gene also deserves the attention of clinicians and inclusion in diagnostic panels. The accumulation of information on PRDM16 changes in patients with CMP will make it possible to clarify the contribution of this gene to primary heart diseases and will develop knowledge about their etiology in general.

Чтобы читать статью войдите с логином и паролем от scardio.ru
Keywords
For citation
Myasnikov R.P., Bukaeva A.A., Kulikova O.V., Ershova A.I., Petukhova A.V., Zotova E.D., Meshkov A.N., Mershina E.A., Kiseleva A.V., Divashuk M.G., Pilyus P.S., Kharlap M.S., Mikova V.M., Koretsky S.N., Gandaeva L.A., Sinitsyn V.E., Basargina E.N., Boytsov S.A., Snigir E.A., Akinshina A.I., Kashtanova D.A., Makarov V.V., Yudin V.S., Drapkina O.M. New variant of PRDM16 gene nucleotide sequence in a family with various phenotypic manifestations of the non-compacted myocardium. Russian Journal of Cardiology. 2021;26(1S):4315. (In Russ.) https://doi.org/10.15829/1560-4071-2021-4315
Copy