Новый вариант нуклеотидной последовательности гена PRDM16 в семье с различными фенотипическими проявлениями некомпактного миокарда
Некомпактный миокард левого желудочка (НМЛЖ) — редкая патология, характеризующаяся аномальным строением миокарда с выраженным некомпактным слоем и повышенной трабекулярностью [1]. В последние годы в связи с улучшением методов визуализации частота обнаружения НМЛЖ существенно возрастает, однако наряду с этим растет и сложность корректной клинической интерпретации данного феномена. Спектр состояний, при которых регистрируется НМЛЖ, варьируется от первичных кардиомиопатий (КМП) и других врожденных заболеваний, ведущих к тяжелой сердечной недостаточности (СН) и требующих радикального лечения, до случайных находок у людей, не предъявляющих жалоб (к примеру, спортсменов и беременных женщин) [2]. В связи с этим оценка морбидности НМЛЖ, определение его роли в развитии сердечной декомпенсации и прогноз заболевания в каждом конкретном случае возможны только после комплексного анализа клинической картины, семейного и генетического анамнеза [3][4].
По актуальным оценкам, не менее половины случаев НМЛЖ имеют наследственную природу; примерно в 30% случаев удается обнаружить патогенный или потенциально патогенный генетический вариант методами ДНК-диагностики [1]. Генетическая детерминированность, т.е. наличие у пациента генетических вариантов, опосредующих развитие НМЛЖ, классифицируется как независимый фактор риска фатальных сердечно-сосудистых событий и неблагоприятного прогноза КМП [2]. В связи с этим пациентам с установленным диагнозом НМЛЖ рекомендовано генетическое тестирование [5]. К настоящему моменту не менее 80 генов ассоциированы с развитием НМЛЖ, и этот список расширяется по мере накопления данных высокопроизводительного секвенирования (NGS). Хотя существенная доля выявляемых у пациентов с НМЛЖ вариантов приходится на гены саркомерных белков миокарда, связанные также с развитием различных типов первичных КМП (в частности, MYH7, MYBPC3, TTN), общий спектр генетических находок при НМЛЖ намного шире и включает большое количество вариантов в генах, кодирующих разнообразные факторы клеточного развития и дифференцировки, в т.ч. факторы эмбриогенеза миокарда и пр. Эти данные подкрепляют гипотезу об эмбриональном происхождении НМЛЖ, однако для уточнения роли таких генов и повышения доказательности их связи с развитием НМЛЖ необходимо больше данных о корреляциях “генотип-фенотип”.
В данной статье представлена семья с различными фенотипическими проявлениями НМЛЖ при наличии одного и того же варианта в гене, кодирующем транскрипционный фактор, ответственный за подавление экспрессии генов, участвующих в эмбриональном развитии, после рождения (рис. 1).

Рис. 1. Родословная.
Описание клинического случая
Пробанд — женщина 33 лет, наблюдающаяся у кардиолога в ФГБУ НМИЦ ТПМ Минздрава России. Телосложение — нормостеническое.
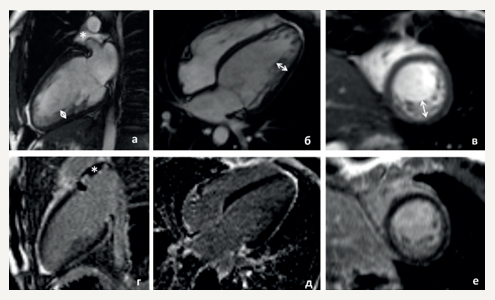
В 27 лет во время первой беременности стали беспокоить перебои в работе сердца. В дальнейшем появилось ощущение нехватки воздуха, головокружение. В октябре 2015г проходила обследование в ФГБУ НМИЦ ТПМ. В анализах крови все стандартно исследуемые показатели были в пределах нормальных значений. При суточном мониторировании электрокардиограммы по Холтеру (ХМ-ЭКГ) была выявлена редкая желудочковая экстрасистолия (ЖЭС). При эхокардиографии (ЭхоКГ) выявлено: конечный диастолический размер (КДР) 5,2 см, толщина межжелудочковой перегородки (ТМЖП) 0,8 см, фракция выброса (ФВ) левого желудочка (ЛЖ) 47%, признаки НМЛЖ в области верхушки и боковой стенки (критерии Chin, Stollberger, Jenni). При магнитно-резонансной томографии (МРТ) сердца с контрастированием (рис. 2) зарегистрирована магнитно-резонансная картина синдрома некомпактного миокарда без дилатации полостей, ФВ 45%. Были назначены бета-адреноблокаторы, ингибиторы ангиотензинпревращающего фермента, антагонисты минералокортикоидных рецепторов. После госпитализации пациентка чувствовала себя удовлетворительно, препараты постоянно не принимала. Ухудшение состояния с весны 2017г, когда возобновились перебои в работе сердца, появилась слабость. По данным ХМ-ЭКГ на фоне бисопролола 2,5 мг/сут., выявлен синусовый ритм с частотой сердечных сокращений (ЧСС) 46-78-144 в мин, 6640 одиночных ЖЭС, 10638 бигеминий, 40 парных ЖЭС, паузы не зарегистрированы. По данным ЭхоКГ, КДР 5,3 см, ТМЖП 0,8 см, ФВ 38%, диастолическая дисфункция 2 типа, признаки НМЛЖ в области верхушки и боковой стенки (критерии Chin, Jenni, Stollberger [6][7][8]). На фоне коррекции терапии: увеличения дозы бисопролола, добавления спиронолактона и периндоприла, состояние пациентки стабилизировалось. При ежегодном динамическом наблюдении, по данным ЭхоКГ, сохраняются нормальные размеры камер сердца и ФВ 45%.

Рис. 2. МРТ сердца пробанда (II-2). (а-в) кино-режим, SSFP-последовательность: а — длинная ось 2-х камерная проекция, б — длинная ось 4-х камерная проекция, в — короткая ось, (г-е) — отсроченное контрастирование, IR-последовательность с подавлением сигнала от миокарда. Участки интрамиокардиального фиброза, рубцового и поствоспалительного поражения миокарда отсутствуют.
Примечания: стрелками указано повышение трабекулярности миокарда ЛЖ в средних боковых и нижнем сегментах; толщина некомпактного слоя 12-15 мм при толщине компактного 5 мм; * — небольшое количество свободной жидкости в верхнем завороте перикарда.
Диагноз НМЛЖ пробанду был установлен на основании ЭхоКГ критериев некомпактного миокарда [9] и был подтвержден на основании МРТ критериев (Jacquier и Petersen) [10][11].
Фенотипический каскадный скрининг
Родословная и клинические данные о родственниках пробанда представлены на рисунке 1 и в таблице 1.
Таблица 1
Клинические данные

Сокращения: ВПС — врожденный порок сердца, МРТ — магнитно-резонансная томография, НМЛЖ — некомпактный миокард левого желудочка, НРС — нарушения проводимости сердца, ТЭО — тромбоэмболические осложнения, ФВ — фракция выброса, ХСН — хроническая сердечная недостаточность, ЭхоКГ — эхокардиография.
Родному брату пробанда 38 лет был проведен кардиологический скрининг, по результатам которого данных за наличие НМЛЖ не получено.
Сводному брату пробанда по материнской линии 25 лет, нормального телосложения (рост 160 см, вес 60 кг), было проведено комплексное кардиологическое обследование. В стандартных анализах все показатели в пределах нормальных значений. По данным ЭхоКГ, КДР 4,9 см, ТМЖП 1,0 см, ФВ 53%, признаки синдрома некомпактного миокарда в области верхушки, боковой и задней стенок (критерий Stollberger). По данным МРТ сердца (рис. 3), камеры сердца не расширены, сократимость миокарда ЛЖ не снижена, участков фиброза, рубцового и поствоспалительного поражения миокарда не выявлено, строение миокарда обычное, несколько повышена трабекулярность среднего и верхушечного сегментов боковой и задней стенок ЛЖ.

Рис. 3. МРТ сердца сводного брата пробанда (II-4). (а-в) кино-режим, SSFP-последовательность: а — длинная ось 2-х камерная проекция, б — длинная ось 4-х камерная проекция, в — короткая ось, (г-е) — отсроченное контрастирование, IR-последовательность с подавлением сигнала от миокарда. Участки интрамиокардиального фиброза, рубцового и поствоспалительного поражения миокарда отсутствуют.
Примечание: * — повышенная трабекулярность в области верхушки ЛЖ за счет “рассыпного” строения папиллярных мышц.
Мать пробанда, 58 лет. Во время третьих родов отмечалось повышение артериального давления (АД) до 160 и 100 мм рт.ст. В дальнейшем не обследовалась, АД не измеряла. С 50 лет при периодическом измерении АД фиксировала цифры 160-180 и 100- 110 мм рт.ст., гипотензивные препараты не принимала. Впервые прошла обследование в НМИЦ ТПМ в возрасте 55 лет. По данным ЭхоКГ выявлены признаки некомпактного миокарда, ТМЖП 1,2 см, в выносящем тракте 1,4 см, задняя стенка ЛЖ 1,5 см, КДР 5,4 см, выраженная трабекулярность миокарда ЛЖ, особенно в области заднебоковой стенки ЛЖ. Миокард ЛЖ представляет собой губчатую, ячеистую структуру с межтрабекулярными лакунами, прокрашиваемыми при цветовом допплеровском картировании. Соотношение компактного (6 мм) и некомпактного (18) слоев миокарда в области задней стенки ЛЖ — 3,0 (выраженная степень некомпактности миокарда по критериям Chin, Stollberger, Jenni). На электрокардиограмме синусовый ритм с ЧСС 65 уд./мин, электрическая ось сердца отклонена влево, полная блокада левой ножки пучка Гиса. При МРТ сердца с контрастированием (рис. 4) выявлена асимметричная гипертрофия миокарда ЛЖ (толщина базальных переднего и переднеперегородочного сегментов 13-14 мм, толщина других базальных и средних сегментов не превышает 7-9 мм, верхушечных — 4-6 мм), папиллярные мышцы гипертрофированы, толщиной 9-12 мм, отмечается их “рассыпной” тип строения, что приводит к выраженной трабекулярности в области верхушки и по переднебоковой стенке ЛЖ с толщиной некомпактного слоя 10-20 мм при толщине компактного 4-7 мм. Масса некомпактного миокарда составляет 16% от массы компактного миокарда. После проведенного обследования были назначены ингибиторы ангиотензинпревращающего фермента, бета-адреноблокаторы, которые пациентка принимает нерегулярно. В настоящее время привычное АД 160-170 и 90 мм рт.ст.

Рис. 4. МРТ сердца матери пробанда (I-4). (а-в) кино-режим, SSFP-последовательность:
а — длинная ось 2-х камерная проекция, б — длинная ось 4-х камерная проекция, в — короткая ось, (г-е) — отсроченное контрастирование, IR-последовательность с подавлением сигнала от миокарда. Участки интрамиокардиального фиброза, рубцового и поствоспалительного поражения миокарда отсутствуют.
Примечания: стрелками указан гипертрофированный переднеперегородочный сегмент (толщиной 14 мм); * — повышение трабекулярности миокарда ЛЖ, в средних боковых сегментах толщина некомпактного слоя составляет 15 мм при толщине компактного слоя 6 мм; ? — небольшое количество жидкости в полости перикарда со стороны правых отделов.
Отец пробанда умер внезапно в возрасте 35 лет (злоупотреблял алкоголем).
Дочери пробанда 4 года. Девочка с 3-х месяцев не прибавляла в весе, тогда же появилась одышка, тахипноэ. При плановом осмотре педиатра был заподозрен врожденный порок сердца, в связи чем проходила стационарное лечение: по данным ЭхоКГ ФВ 35%, повышенная трабекулярность ЛЖ, лево-правый сброс в центральной части межпредсердной перегородки размером 3 мм. По данным ультразвукового исследования почек, увеличение обеих почек. Была назначена терапия преднизолоном 5 мг/сут., спиронолактоном 12,5 мг/сут., дигоксином 0,05 мг/сут. В связи с отсутствием положительной динамики для верификации диагноза была направлена в ФГАУ “НМИЦ ЗД” Минздрава России. По данным ЭхоКГ, выраженная дилатация левых отделов сердца (КДР 3,8, КСР 3,0 см), ФВ 36%, НМЛЖ, открытое овальное окно 1,5 мм. ХМ-ЭКГ: синусовый ритм с ЧСС 80-122-172, парциальный феномен предвозбуждения, единичная ЖЭС. На фоне подобранной терапии состояние улучшилось. Постоянно принимает амиодарон 50 мг/сут., диакарб по схеме, дигоксин 0,02 мг/сут., каптоприл по 2,5 мг 3 раза/сут., фуросемид 3 мг/сут., карведилол по 0,78 мг 2 раза/сут. В настоящее время явления некомпактного миокарда практически купированы. Ребенок развивается соответственно возрасту.
В таблицах 2 и 3 объединены результаты визуализирующих методов обследования пробанда и ее родственников.
Таблица 2
Выраженность некомпактного миокарда, по данным МРТ сердца, у пробанда и его родственников

Сокращение: нм/к — отношение некомпактного слоя к компактному.
Таблица 3
Показатели МРТ сердца у пробанда и его родственников

Сокращения: КДО — конечный диастолический объем, НМ — некомпактный миокард, Нм/массе миокарда — отношение массы НМ к индексированной массе, Нм/к — отношение некомпактного слоя к компактному, ФВ ЛЖ — фракция выброса левого желудочка.
Генетический каскадный скрининг
Пробанду и всем родственникам первой степени родства был проведен молекулярно-генетический анализ.
Секвенирование полного генома и биоинформатический анализ
ДНК была выделена из образцов цельной крови с помощью набора QIAamp DNA Blood Mini Kit (Qiagen, Германия). Библиотека WGS была подготовлена с использованием набора Nextera DNA Flex kit (Illumina, США) в соответствии с инструкциями производителя. Средняя глубина покрытия при секвенировании (150 п.н., парные чтения) составила от 30X и более. Чтения были выровнены на референсный геном (GRCh38), а небольшие варианты были найдены с помощью платформы Dragen Bio-IT (Illumina, США) и уточнены с использованием GLnexus [12]. Аннотация проводилась с использованием Ensembl VEP [13]. Для клинической интерпретации были отобраны варианты нуклеотидной последовательности в генах, ассоциированных с развитием НМЛЖ по имеющимся литературным данным, с частотами <0,5% в базе данных gnomAD. Оценка патогенности вариантов проводилась в соответствии с критериями, изложенными в актуальном отечественном руководстве по интерпретации данных NGS [14]. Верификацию находок выполняли путем прямого двунаправленного секвенирования по Сенгеру.
Результаты молекулярно-генетического исследования
В результате молекулярно-генетического исследования выявлена ранее не описанная однонуклеотидная делеция в гене PRDM16, приводящая к сдвигу рамки считывания в 9 экзоне и образованию преждевременного стоп-кодона (NM_022114.4: c.2436delT; NP_071397.3: p.Ala813ProfsTer58) (рис. 5). Вариант подтвержден у пробанда, ее дочери, матери и сводного брата по материнской линии. На основании актуальных критериев патогенности находка классифицирована как патогенный вариант (V класса патогенности). У родного брата пробанда указанный вариант не обнаружен.

Рис. 5. Экзонная структура гена PRDM16. Корректно считываемая последовательность отмечена зеленым, участок считывания в неправильной рамке — голубым, нечитаемый участок — серым. Желтый маркер отмечает мутацию, звездочка — преждевременный стоп-кодон.
Примечание: Цветное изображение доступно в электронной версии журнала.
Обсуждение
В данной работе представлена семья с различными фенотипами НМЛЖ. Наиболее тяжелое течение заболевания отмечается у дочери пробанда, у которой диагностирован дилатационный тип НМЛЖ с тяжелым течением СН. У пробанда выявлен изолированный НМЛЖ с незначительным снижением систолической функции ЛЖ до 46%. В клинической картине преобладают симптомные нарушения ритма сердца в виде частой ЖЭС, без пробежек желудочковой тахикардии. У матери пробанда обращает на себя внимание наличие гипертрофии миокарда на фоне НМЛЖ, что, в свою очередь, может быть обусловлено повышением АД. Учитывая бессимптомное течение, фактическое начало артериальной гипертонии остается неизвестным, в связи с чем исключать гипертонию как причину гипертрофии миокарда на фоне изначально скомпрометированного миокарда не представляется возможным.
У всех членов семьи с фенотипом НМЛЖ (у пробанда, ее дочери, матери и сводного брата по материнской линии) выявлен патогенный вариант PRDM16, приводящий к утрате копии гена. PRDM16 кодирует белок, представляющий собой фактор транскрипции, содержащий домены типа “цинковые пальцы”. PRDM16 образует комплексы с различными транскрипционными кофакторами и модуляторами хроматина. В зависимости от биологической задачи он может стимулировать или подавлять тканеспецифичную экспрессию генов. Роль PRDM16 широко изучалась в жировой ткани [15]. Одновременно с этим показана экспрессия PRDM16 в кардиомиоцитах мыши и человека [16][17].
Связь гена PRDM16 с развитием НМЛЖ была впервые предположена в исследовании Arndt A-K, et al. в 2013г [16] на основании частой встречаемости дилатационной КМП и НМЛЖ у носителей делеции участка 1p36 на 1 хромосоме, включающего данный ген. Последующие исследования подтвердили роль вариантов, приводящих к утрате копии PRDM16, в развитии НМЛЖ и дилатационной КМП у детей [18]. В экспериментах на нокаутированных по гену PRDM16 мышах была показана роль PRDM16 в развитии гипертрофической КМП [17][19]. В норме PRDM16 играет протективную роль, но отсутствие PRDM16, согласно Cibi и др. (2020), приводит к гипертрофии миокарда, чрезмерному фиброзу желудочков, дисфункции митохондрий и нарушению метаболических процессов в клетке, что способствовало развитию СН во взрослом возрасте [17]. Эффект опосредован реактивацией генетической программы эмбриогенеза у взрослых мышей, т.е. сохранением высокой активности генов, участвующих в эмбриональном развитии, но находящихся в дезактивированном состоянии после рождения. У нокаутированных по PRDM16 мышей наблюдалась активация гипертрофических генов NPPA, NPPB, MYH7, MYL14, повышение экспрессии генов, участвующих в развитии фиброза (TGFb2, CTGF, TIMP4, LTBP2), генов, вовлеченных в метаболизм углеводов (BDH1, PDK4, GLUT1, NR4a1, HMGCS2, PPARG), снижение экспрессии генов, участвующих в окислении жирных кислот (FASN, CD36, SCD1, SCD2, ADIPOQ), генов, ответственных за функцию митохондрий (mt-ND4, GPAM, UCP3, DLAT, MTHFD2), метаболизм железа (TFRC, HAMP, ALAS1, ALAS2, LCN2) и др. [17]. В работе Cibi DM, et al. (2020) также показано, что гипертрофическая КМП могла развиваться у молодых мышей, но в ответ на метаболический стресс [17].
Однако клинически значимых находок для вариантов в гене PRDM16 пока слишком мало, чтобы уверенно оценивать его вклад в этиологию заболевания. В курируемой базе данных Clinical Genome Resource (www.clinicalgenome.org) уровень доказательности связи “ген-болезнь” для PRDM16 в настоящее время обозначен как “ограниченный”. Важность продукта гена PRDM16 для нормального развития кардиомиоцитов была показана на животных моделях [16], однако для однозначного вывода о связи этого гена с НМЛЖ и первичными КМП имеющихся данных недостаточно.
Косегрегация варианта с симптомами в рамках семьи является одним из ключевых аргументов в пользу связи этого варианта с развитием заболевания и основанием для повышения класса патогенности вариантов с неоднозначной интерпретацией. Мы полагаем, что наша находка подкрепляет имеющиеся данные о роли PRDM16 в патогенезе НМЛЖ и свидетельствует о целесообразности включения данного гена в генетические панели для диагностики КМП. Однако остается открытым вопрос разнообразия клинических проявлений в рамках одной семьи. Вопервых, согласно данным литературы, сами варианты PRDM16 могут быть ассоциированы как с диастолической, так и гипертрофической КМП (в эксперименте на животных). Объяснения данному факту в литературе отсутствуют. Во-вторых, тяжелый фенотип у дочери пробанда позволяет ожидать присутствие второго патогенного варианта, утяжеляющего течение КМП, однако иных потенциально значимых вариантов у ребенка выявлено не было. В-третьих, раннее и более тяжелое течение НМЛЖ у дочери пробанда может быть следствием метаболических нарушений в неонатальном периоде. Четких анамнестических данных в пользу данного предположения нет, однако стабилизация клинического течения и нормальное развитие ребенка в течение последних трех лет говорят в пользу перенесенного стрессового фактора, приведшего к метаболическим нарушениям и запустившего эпизод значимой активации генов, ответственных за эмбриональное развитие, которая не была предотвращена вследствие отсутствия экспрессии PRDM16.
Заключение
В данной статье представлена семья с различными фенотипическими проявлениями НМЛЖ при наличии одного и того же ранее не описанного варианта в гене PRDM16, кодирующем транскрипционный фактор, ответственный за подавление в постнатальном периоде экспрессии генов, участвующих в эмбриональном развитии. Сегрегация варианта с симптомами в трех поколениях семьи свидетельствует в пользу связи выявленного варианта с развитием НМЛЖ. На текущий момент ввиду малой информированности о роли гена PRDM16 в развитии патологии, влиянии вариантов в нем на течение заболевания и, тем более, о путях персонализации терапии для носителей таких вариантов, мы не можем какимлибо образом менять тактику ведения пациентов на основании нашей находки. Однако в условиях все более широкого внедрения высокопроизводительных методов молекулярной генетики в схему наблюдения пациентов с первичными заболеваниями сердца сообщение подобных находок, не несущих явной практической пользы на данном этапе, представляется необходимым для привлечения внимания к недостаточно изученным генам-кандидатам НМЛЖ. Текущая схема ДНК-диагностики КМП в клинике зачастую предполагает фокусировку внимания на хорошо изученных генах. Обнаруженный нами вариант и его косегрегация с признаками НМЛЖ в трех поколениях, вкупе с предыдущими литературными данными о находках в PRDM16, свидетельствуют о том, что этот ген также заслуживает внимания клиницистов и включения в диагностические панели. Накопление информации об изменениях в PRDM16 у пациентов с КМП позволит уточнить вклад этого гена в спектр причин первичных заболеваний сердца и будет способствовать расширению знаний об их этиологии в целом.

Чтобы читать статью войдите с логином и паролем от scardio.ru
Ключевые слова
Для цитирования
Мясников Р.П., Букаева А.А., Куликова О.В., Ершова А.И., Петухова А.В., Зотова Е.Д., Мешков А.Н., Мершина Е.А., Киселева А.В., Дивашук М.Г., Пилюс П.С., Харлап М.С., Микова В.М., Корецкий С.Н., Гандаева Л.А., Синицын В.Е., Басаргина Е.Н., Бойцов С.А., Снигирь Е.А., Акиньшина А.И., Каштанова Д.А., Макаров В.В., Юдин В.С., Драпкина О.М. Новый вариант нуклеотидной последовательности гена PRDM16 в семье с различными фенотипическими проявлениями некомпактного миокарда. Российский кардиологический журнал. 2021;26(1S):4315. https://doi.org/10.15829/1560-4071-2021-4315
Скопировать