Гипертрофическая кардиомиопатия в сочетании с синдромом Бругада
Аннотация
Введение. Сочетание синдрома Бругада и гипертрофической кардиомиопатии является крайне редким, характеризуется высоким риском развития неблагоприятного исхода и вызывает множество вопросов относительно тактики ведения таких пациентов.
Краткое описание. Представлен клинический случай девочки с сочетанием фенотипа гипертрофической кардиомиопатии и электрокардиографического (ЭКГ) паттерна Бругада. Заболевание дебютировало в 4 года, когда при обследовании выявлены гипертрофия миокарда левого желудочка и преходящий ЭКГ-паттерн по типу «свода» в правых грудных отведениях. ЭКГ изменения изначально были расценены как вторичные — на фоне гипертрофической кардиомиопатии. В возрасте 7 лет, после появления специфических жалоб и получения результатов полногеномного секвенирования, был верифицирован синдром Бругада. Сочетание двух заболеваний потребовало коррекции терапии и тактики ведения.
Дискуссия. Наш пример представляет сочетание генетически верифицированных гипертрофической кардиомиопатии и синдрома Бругада. В мировой литературе у детей подобные случаи ранее не описаны. Комбинация двух редких потенциально летальных заболеваний требует от лечащего врача крайне внимательного подхода к подбору медикаментозной терапии, особенно бета-адреноблокаторов, динамическому наблюдению и тактике дальнейшего ведения.
Введение
Гипертрофическая кардиомиопатия (ГКМП) — генетически обусловленное заболевание, характеризующееся гипертрофией миокарда, чаще асимметричного характера, не связанной с повышением постнагрузки на миокард, клапанной патологией, пороками сердца и сосудов [1]. Это этиологически гетерогенное заболевание, только у 30-60% пациентов удается определить каузативные варианты [2]. Более 50% случаев ассоциировано с мутациями в генах, кодирующих саркомерные белки — это саркомерная или первичная ГКМП. Наиболее часто встречаются мутации в генах MYH7 и MYBPC3, реже в TNNT2, TNNI3 и ACTC1 [3]. В отдельную группу выделяют фенокопии ГКМП (болезни накопления, нейромышечные заболевания, митохондриальные болезни, RAS-опатии) [4]. ГКМП имеет неблагоприятный прогноз и высокую летальность. В детском и молодом возрасте смерть в большинстве случаев наступает внезапно в результате развития жизнеугрожающих аритмий (ЖУА) [5]. При обследовании пациентов с ГКМП встречаются различные электрокардиографические (ЭКГ) феномены: нарушение внутрижелудочковой проводимости, ЭКГ-паттерн Бругада, феномен преэкзитации, удлинение интервала QT [6]. В таких случаях дифференциальный диагноз сложен и требует углубленного обследования для определения генеза изменений электрической активности миокарда — первично, вследствие нарушения работы ионных каналов или вторично, на фоне изменения морфологической структуры миокарда.
Синдром Бругада (СБ) — это первичное электрическое заболевание сердца, которое характеризуется специфическим ЭКГ-паттерном в виде нарушения проводимости по правому желудочку (ПЖ), элевацией сегмента ST в правых грудных отведениях и высоким риском внезапной сердечной смерти (ВСС) из-за желудочковых аритмий [7][8]. Сроки манифестации СБ варьируют от раннего детства до пожилого возраста. Наиболее часто развернутая клиническая картина СБ проявляется в возрасте 30-45 лет, чаще у лиц мужского пола, а средний возраст ВСС составляет 41±15 лет [9]. Для верификации СБ используется Шанхайская балльная система, в которой оцениваются ЭКГ-критерии, клинические признаки, семейный анамнез, результаты генетического исследования. Выделяют три ЭКГ-типа СБ (первый — выраженный подъём точки J, изменение сегмента ST сводчатое и инверсия T-волны в отведениях V1 и V2; второй — седловидный подъём сегмента ST более чем на 1 мм; третий — подъём сегмента ST менее чем на 1 мм)1 [10]. В настоящее время основной причиной СБ принято считать мутации в гене SCN5A, которые обнаруживаются в 20-30% случаев. С этим заболеванием связаны ещё 23 гена, но в совокупности они встречаются лишь в 5-10% случаев. Таким образом, ~70% случаев СБ остаются необъяснёнными. На сегодняшний день рекомендованный генетический скрининг ограничивается геном SCN5A [10][11].
Точная распространенность ГКМП среди пациентов детского возраста неизвестна. На основании последнего исследования о частоте первичных кардиомиопатий среди детей (0,002%), предполагаемая распространенность ГКМП составляет 0,0007-0,001%, что в сравнении со взрослыми значимо ниже (0,2%) [12]. СБ также является крайне редким в детской популяции и имеет крайне низкую распространенность (0,0098%), в сравнении со взрослыми частота встречаемости в 10-50 раз ниже [13]. Сочетание генетически верифицированных ГКМП и СБ в литературе не описано — распространенность неизвестна. Представляем клиническое наблюдение девочки с определенными мутациями в генах MYH7, MYL3 и SCN5A, с сочетанием фенотипа ГКМП, являющейся наиболее частой причиной ВСС в детском и молодом возрастах, и СБ, ответственного за 4-12% всех случаев ВСС [13][14].
Клинический случай
Ребенок от 3 беременности, протекавшей на фоне угрозы прерывания на 8 нед., стресса вследствие внезапной смерти старшего ребёнка. Роды вторые, срочные, стремительные. Массо-ростовые показатели при рождении в пределах нормальных значений. Раннее физическое и нервно-психическое развитие — в соответствии с возрастом.
Учитывая отягощенный семейный анамнез по ГКМП (отец пациентки) и ВСС (смерть сибса во сне в возрасте 11 мес.) (рис. 1), в 2015г при рождении ребенка проведено прицельное инструментальное обследование — структурной патологии не выявлено. После диагностирования у младшего брата в 2018г обструктивной ГКМП у членов семьи взят биоматериал с целью проведения молекулярного-генетического исследования в объёме полноэкзомного секвенирования. По результатам исследования был выявлен гетерозиготный вариант в гене МYH7, приводящий к аминокислотной замене p.Asn897His, у пробанда, отца и младшего брата.

Рис. 1. Генеалогическое древо: A1 — ВСС во сне в 11 мес. жизни (на вскрытии без патологии сердечно-сосудистой системы); A2 — пробанд; А3 — обструктивная ГКМП, гетерозиготная мутация в гене MYН7 (миосептоэктомия в 2022г); А4 — не обследован; B1 — ГКМП, мутации в генах MYH7, MYL3, синкопальные состояния в анамнезе; B2 — здорова. Дополнительно: ВСС по линии отца — у двоюродных братьев и сестер (в т.ч. во сне).
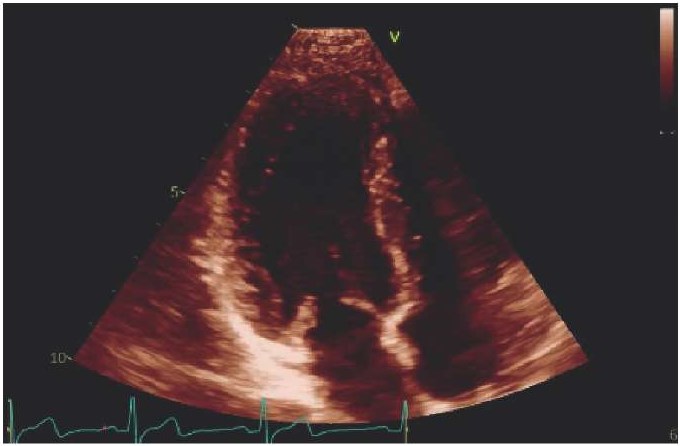
В 2019г ребенок впервые госпитализирован в детское кардиологическое отделение (Временная шкала). При физикальном осмотре грудная клетка без изменений, аускультативно тоны сердца ясные ритмичные, шум не выслушивается. По данным трансторакальной эхокардиографии выявлена асимметричная гипертрофия миокарда левого желудочка с максимальной толщиной миокарда до 11 мм (3,78 Z-score, N до 2 Z-score) (рис. 2). На ЭКГ зарегистрированы нарушение проведения по правой ножке пучка Гиса и высокие зубцы Т в отведениях V3-V4, которые были расценены как вторичные изменения на фоне структурной патологии миокарда (рис. 3).

Временная шкала
Сокращения: ДКО — детское кардиологическое отделение, ГКМП — гипертрофическая кардиомиопатия, ОРВИ — острая респираторная вирусная инфекция, ССС — сердечно-сосудистая система, ЭКГ — электрокардиография, ЭФИ — электрофизиологическое исследование, ЭхоКГ — эхокардиография.

Рис. 2. Трансторакальная эхокардиография, 4-х камерная проекция.

Рис. 3. Поверхностное 12-канальное ЭКГ.
Далее ребенок наблюдался ежегодно — состояние без отрицательной динамики по данным лабораторных и инструментальных методов обследования. В 2020г проведен пересмотр результатов полноэкзомного секвенирования — дополнительно выявлена гетерозиготная мутация в гене MYL3, приводящая к аминокислотной замене p.Ala57Asp. Вариант также обнаружен у отца пробанда.
При обследовании в 2021г по результатам трансторакальной эхокардиографии толщина миокарда без изменения (11 мм/3,05 Z-score), однако отмечена тенденция к дилатации левого предсердия (индекс объема 33 мл/м²). В связи с чем с кардиопротективной целью ребенку были назначены спиронолактон и пропранолол. При стратификации риска ВСС по шкале HCM Risk-Kids определен низкий риск аритмических событий, показаний для имплантации кардиовертера-дефибриллятора нет.
В конце 2022г на фоне острой респираторной инфекции, со слов матери, ребенок предъявлял жалобы на ухудшение самочувствия в виде повышенной утомляемости, ощущений сердцебиения. По данным суточного ЭКГ-мониторирования на фоне терапии пропранололом выявлены преходящие ЭКГ паттерны по типу «свода» и «седла» в отведениях V1, V2 в виде полной блокады правой ножки пучка Гиса и элевации сегмента ST.
В 2023г получены результаты полногеномного секвенирования — ранее описанные мутации выявлены и подтверждены, а также выявлен не описанный в литературе вариант в гетерозиготном состоянии в гене SCN5A, приводящий к аминокислотной замене р.Аsр1041Нis. Вариант также обнаружен у матери пробанда (спонтанный ЭКГ-паттерн Бругада отсутствует).
С учётом выявленной мутации в гене SCN5A, регистрации преходящих ЭКГ-паттернов Бругада, внезапной смерти родственника 1 линии родства — было принято решение о проведении медикаментозной пробы с использованием антиаритмического препарата 1А класса новокаинамида в условиях отделения реанимации. Доза препарата выбрана из расчёта 10 мг/кг — 2,9 мл внутривенно капельно в течение 10 мин.
Исходно на ЭКГ регистрировался синусовый ритм, комплекс QRS в отведениях V1-V2 имел морфологию RSr’, также в этих отведениях был отрицательный зубец Т. На 4 мин введения препарата (введено ≈1,16 мл новокаинамида) отмечено появление косонисходящей элевации сегмента ST в правых грудных отведениях, а также удлинение интервала QT в правых прекордиальных отведениях V1-V2 и альтернация зубца Т. Через 6,5 мин (введено ≈1,9 мл новокаинамида) зарегистрированы такие ЭКГ-паттерны Бругада 1 типа, как расположение самой высокой точки комплекса QRS-ST выше 2 мм над изолинией, морфология сегмента ST в виде свода с последующим отрицательным симметричным зубцом T, снижение сегмента ST после пика QRS происходит медленно (менее 0,4 мВ за 40 мс), индекс Коррадо >1 (соотношение между пиковой высотой комплекса QRS-ST и пиком сегмента ST через 80 мс), длительность комплекса QRS в отведениях V1-V2 больше, чем в средних и левых прекордиальных отведениях, появление фрагментации QRS-комплекса в отведениях V1-V2 (рис. 4), в связи с чем введение препарата прекращено, проба завершена. Примечательный факт, что медикаментозно спровоцированные ЭКГ-изменения регистрировались на протяжении двух суток после проведения пробы с восстановлением исходных показателей на третьи сутки.

Рис. 4 А. ЭКГ. Проба с новокаинамидом, исход.

Рис. 4 Б. ЭКГ. Проба с новокаинамидом, 4 мин.

Рис. 4 В. ЭКГ. Проба с новокаинамидом, 6,5 мин.
Учитывая положительный результат медикаментозной пробы, согласно клиническим рекомендациям 2022г Европейской ассоциации кардиологов [15], у ребенка выявлены показания для проведения электрофизиологического исследования с целью стратификации риска ВСС. По результатам: антеградно — AH-интервал 65 мс, HV-интервал 75 мс, точка Венкебаха 215 имп/мин, эффективный рефрактерный период (ЭРП) атриовентрикулярного узла меньше ЭРП предсердий и составил 260 мс; ретроградно-вентрикулоатриальная диссоциация, ЭРП ПЖ 240 мс; при проведении учащающей и программированной стимуляции из выводного отдела ПЖ и верхушки ПЖ с 1, 2 и 3 экстрастимулами тахисистолических нарушений ритма не индуцировано.
Принимая во внимание генетически подтвержденный СБ, опираясь на анализ литературы и рекомендаций, проведена смена пропранолола на метопролол [16]. Стратификация риска развития ЖУА проведена в двух направлениях. У пациентки с ГКМП имелся один большой фактор риска ВСС (отягощенная наследственность по ВСС в молодом возрасте), расчетный 5-летний риск ВСС (HCM Risk-Kids) составлял 3,19%, что указывает на низкий риск возникновения аритмических событий. У больной с СБ, имеющей нарушения проводимости без клинической симптоматики, на основании шкалы, разработанной Gonzalez Corcia MC, et al. в 2018г вероятность возникновения ЖУА в течение 5 лет составила 0% [17]. Таким образом, показаний для имплантации кардиовертера-дефибриллятора нет.
На основании комплексного обследования у пациентки диагностированы ГКМП, ассоциированная с гетерозиготными вариантами в генах MYH7 и MYL3, и СБ, ассоциированный с гетерозиготным вариантом в гене SCN5A. При динамическом наблюдении в течение 6 лет состояние без отрицательной динамики.
Обсуждение
ГКМП и СБ являются редкими заболеваниями с высоким риском возникновения ЖУА. Их сочетание потенцирует вероятность неблагоприятного исхода, однако предполагаемая частота встречаемости крайне мала. Так, в проанализированной литературе не было обнаружено описаний генетически подтвержденных комбинаций этих двух заболеваний.
Сообщается о 8 случаях регистрации ЭКГ-паттерна Бругада у взрослых больных с ГКМП без доказанной генетической причины СБ. Возраст больных варьировал от 22 до 66 лет, 75% были мужчинами, у 6 была отягощенная наследственность по ВСС (3 из одной семьи) [18]. Из них у 2 неродственных больных была мутация в гене MYBPC3 (c.1458-1G>A), у 4 членов одной семьи определен вариант в гене TPM1 (c.574G>A) и 2 не были генотипированы [19]. Farnè M, et al. сообщили о 4 неродственных взрослых пациентах без фенотипических проявлений ГКМП с наличием вариантов в генах MYH7 или MYBPC3 и ЭКГ-паттерном СБ при отсутствии мутаций, ассоциированных с СБ [20]. В публикации Pappone C, et al. в рамках одной семьи, носителей мутации в гене MYBPC3, фенотипические проявления были различны: у отца ГКМП без ЭКГ-паттерна Бругада, у дочери лекарственно индуцированный ЭКГ-паттерн Бругада, у сына спонтанный ЭКГ-паттерн Бругада [21].
Вышеописанные случаи в совокупности с неясной клинической значимостью выявленной мутации в гене SCN5A (D1041H) и отсутствием специфических нарушений проводимости у матери пробанда, которая также является носителем варианта, не позволяют исключить возможность ассоциации ЭКГ-паттерна Бругада с мутациями в генах саркомерных белков. Так, в работе Baruteau AE, et al. из 442 детей с мутацией в гене SCN5A только 8,6% детей (n=38) имели СБ, из них изолировано от других нарушений ритма и проводимости 21,5% (n=8) [22]. Вместе с тем среди детей с диагностированным СБ в грудном возрасте >90% случаев были обусловлены вариантами в гене SCN5A [23].
В связи с возможностью синергизма вариантов в генах MYH7, MYL3 и SCN5A, относительно риска ВСС, при определении тактики ведения пациента оценивается риск неблагоприятного исхода, ассоциированный и с ГКМП, и с СБ. Стоит отметить, что риск ЖУА при СБ наиболее высок во взрослом возрасте. Это подтверждается в работе Righi D, et al. (n=43), в которой только у 5% детей отмечены ЖУА, против представленной Шишко В. В. и др. частоты возникновения аритмических событий у взрослых, равной 17-62% [24][25]. В то время как при ГКМП риск ВСС наиболее высок в детском и молодом возрастах — частота возникновения ВСС у детей в 2,5-3 раза выше, чем у взрослых [14]. В целом решение об имплантации кардиовертера-дефибриллятора как средства профилактики ВСС является трудным в отношении молодых пациентов и требует взвешенной оценки пользы и риска осложнений.
Одним из факторов, усложняющих ведение данного пациента, является то, что для лечения ГКМП у детей препаратом первого выбора являются β-блокаторы, которых при СБ рекомендовано избегать (brugadadrugs.org). В связи с чем неселективный β-адреноблокатор пропранолол был заменен на селективный β1-адреноблокатор метопролол.
Как пациенту с ЭКГ-паттерном Бругада, нашей больной было рекомендовано соблюдать ряд предосторожностей: раннее снижение температуры тела при возникновении лихорадки любого генеза; избегать приема лекарственных средств, которые могут привести к усилению выраженности изменений комплекса QRS-ST; избегать обильного приема пищи с высоким содержанием углеводов, особенно перед сном [13]. Также рекомендовано ограничение физических и психоэмоциональных нагрузок, учитывая фенотип ГКМП.
Заключение
Таким образом, данный пациент подлежит особо внимательному наблюдению детского кардиолога, тщательному подбору терапии и взвешенной тактике дальнейшего лечения.
Информированное согласие. От законных представителей пациента было получено информированное добровольное согласие.
Отношения и деятельность: все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье.
1. Клинические рекомендации «Синдром Бругада». Ассоциация сердечно-сосудистых хирургов России. 2020 г. https://racvs.ru/clinic/files/2020/brugada.pdf.

Чтобы читать статью войдите с логином и паролем от scardio.ru
Ключевые слова
Для цитирования
Поздняков С.В., Пресова В.В., Егоров Л.В., Калачанова Е.П. Гипертрофическая кардиомиопатия в сочетании с синдромом Бругада. Клинический случай. Российский кардиологический журнал. 2026;31(1S):6579. https://doi.org/10.15829/1560-4071-2026-6579. EDN: UPTALP
Скопировать


