Difficult diagnostics of a rare cause of pulmonary hypertension: a case report
Interest in studying the causes of development and approaches to treatment of pulmonary hypertension (PH) has significantly increased in the last 2 decades. This is due to the expansion of both diagnostic techniques and therapeutic strategies, which contributed to significant progress in improving the quality and duration of life of PH patients. According to comprehensive clinical classification of PH by the European Society of Cardiology (2015), there are 5 main groups [1]:
1) Pulmonary arterial hypertension (PAH);
2) PH due to left heart disease;
3) PH due to lung diseases and/or hypoxia;
4) Chronic thromboembolic PH and other pulmonary artery (PA) obstructions;
5) PH with unclear and/or multifactorial mechanisms.
The last (fifth) group is a fairly heterogeneous set of diseases that are rarely found in the practice of a cardiologist.
The presented paper describes a stepwise diagnostic analysis of an extremely rare case of PH in a patient with myeloproliferative disease.
Case report
Fifty-years-old female patient, was admitted to the clinic with complaints of severe weakness, shortness of breath with minimal exertion and at rest, palpitations, increased blood pressure up to 160-170/95 mm Hg, abdominal distention, joint pain, dry and flaky skin.
The described complaints were present for one year with a progressive decrease in exercise tolerance. Two years ago, the patient was examined by a gynaecologist for uterine fibroids, during which anaemia, leucocytosis, and thrombocytosis were revealed. Within 2 years she lost 18 kg. For about 1 year, there are pains in different joints, severe weakness, and peripheral lymph node enlargement (cervical, occipital, inguinal, axillary nodes). She was consulted by a rheumatologist and haematologist, and foot arthrosis was diagnosed. A hypothesis was made about a lymphoproliferative disease, in connection with which sternal puncture and lymph node biopsy were performed, according to which no signs of acute and chronic hemoblastosis were obtained. Therapy with vitamin B12 and folic acid was prescribed, against which the haemoglobin level increased.
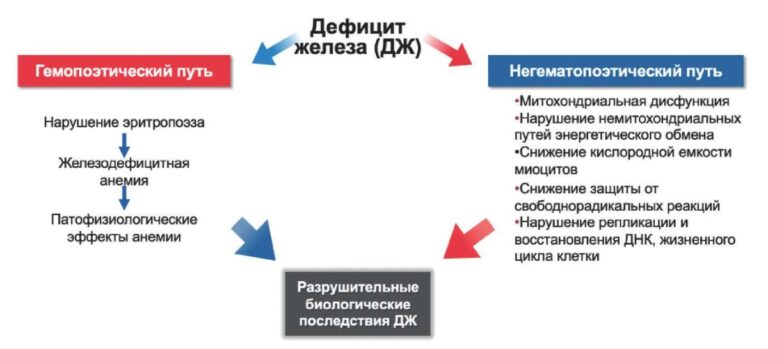
Physical examination. Status: severe. Asthenic type, debilitated. Height — 160 cm, weight — 40 kg. Body mass index — 15,62 kg/m2. Skin: dry; hyperpigmentation, sloughing; moderate acrocyanosis (Figure 1). Submandibular and axillary lymph nodes were enlarged. Visible mucous membranes: pale, cyanotic. Subcutaneous tissue was practically absent. Lower leg and foot swelling; anterior abdominal wall swelling, ascites. Cardiovascular system: regular heart rate (HR) of 98 beats per min. Loud second heart sound at pulmonic area, soft systolic murmur at the apex; pulse of 98 beats per min, regular, weak, blood pressure: 100/60 mm Hg. Respiratory system: chest — right shape; vesicular breathing, weakened; no wheezing; respiratory rate of 20 per minute. Digestive system: tongue — dry, white. The abdomen is enlarged, right upper quadrant discomfort on palpation, free fluid in the abdominal cavity. Liver: enlarged — 2,5-3 cm below the right costal margin; the edge is dense, even, the size of 12?10?9 cm according to Kurlov’s method. The musculoskeletal system: joint deformity (Figure 2). Genitourinary system: Murphy’s punch sign is negative on both sides.
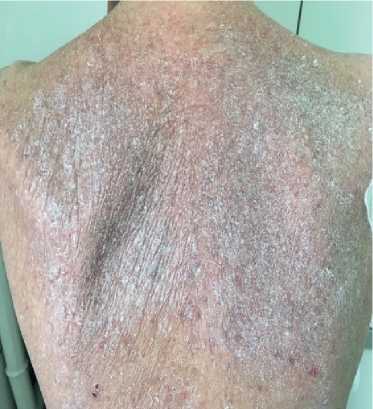
Figure 1. Keratosis of back skin of 50-years-old female patient.
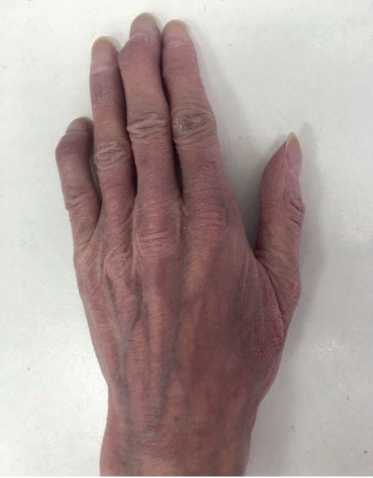
Figure 2. Hand joint deformities of 50-years-old female patient.
Thus, the patient had the following symptom complexes: 1) oedema (ascites, lower limb oedema); 2) hepatomegaly; 3) skin; 4) articular; 5) respiratory failure; 6) lymphadenopathy.
According to diagnostic tests, there were following findings:
Electrocardiography — sinus tachycardia with a heart rate of 100 bpm. Right axis deviation. Low wave voltage. Incomplete right bundle branch block. Signs of right ventricular (RV) overload (Figure 3).
Figure 3. Electrocardiogram upon admission of 50-years-old female patient: sinus rhythm, heart rate of 86 bpm. QT length of 400 ms. Right axis deviation. Low wave voltage. Incomplete right bundle branch block.
Echocardiography — ascending aorta 30 mm. Aortic regurgitation: ++. Left atrium: 29 mm. Left ventricular (LV) end diastolic dimension: 38 mm, interventricular septum (IVS), 9 mm, LV posterior wall, 10 mm. LV ejection fraction: 43%. LV diffuse hypokinesis with paradoxical septal motion. Mitral valve Doppler ultrasound: +. No pronounced response to respiratory phases. Right atrium (RA): 55?58 mm. RV: 32 mm. Right ventricular inflow tract: 53 mm. Diffuse RV hypokinesis. Tricuspid valve (TV) leaflets: sealed, thickened. TV prolapse. TV Doppler ultrasound: +++, (+++/++++). LA: with signs of severe hypertension. Diastolic pressure: 25 mm Hg. Systolic pressure: 90 mm Hg. LA Doppler ultrasound: ++. Inferior vena cava: 20 mm, inadequate response, with spontaneous contrast enhancement effect. Echo-free space along the entire heart perimeter: in front of RV from the epigastric view, 12 mm; above the RV, 5 mm; at the LV apex, 5 mm; behind the lateral wall, 12 mm; behind the LV, 17 mm (Figure 4, 5).
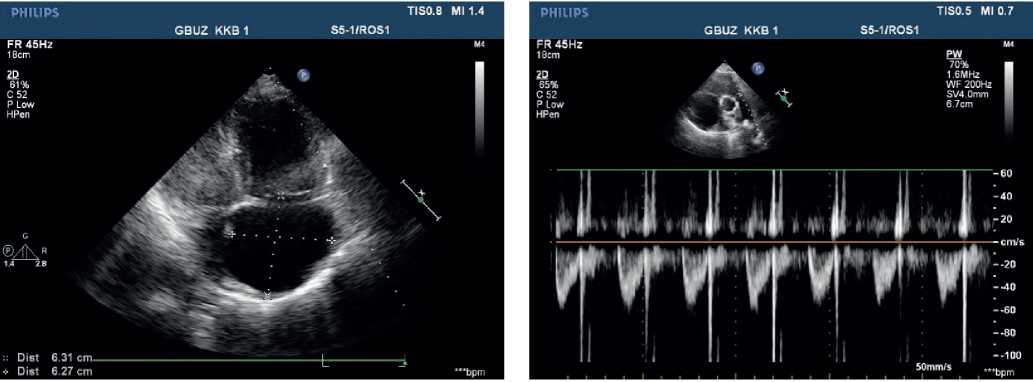
Figure 4, 5. Echocardiography upon admission of 50-years-old female patient: severe PH. Severe TV regurgitation. Right heart dilation.
Chest X-ray — no focal and infiltrative abnormalities. Grade 3 cardiomegaly. Cardiothoracic ratio was 0,52 (Figure 6).
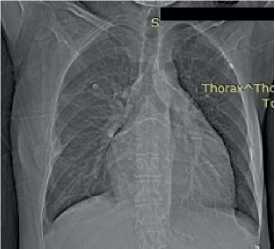
Figure 6. Chest X-ray of 50-years-old female patient. no focal and infiltrative abnormalities. Sinuses are clear. The left heart is enlarged. Cardiothoracic ratio is 0,52.
Based on the above data, the differential diagnosis included: cancer with paraneoplastic syndrome, cardiovascular disease, pulmonary disorder with development of pulmonary heart disease and heart failure. A combination of several diseases was not ruled out.
The patient underwent esophagogastroduodenoscopy, colonoscopy, transvaginal ultrasound. There were no data suggestive of cancer.
When analysing the echocardiography, attention was drawn to the high PA pressure and right heart enlargement without left heart structural pathology.
According to clinical guidelines for the diagnosis and treatment of patients with PH, PH is most common due to left heart disease [1].
To rule out the left heart disease as a possible cause of PH, a Swan-Ganz catheter was placed, according to which the pulmonary wedge pressure was 13 mm Hg, PA systolic pressure — 48 mm Hg, and pulmonary vascular resistance — 380 mm. Pulmonary vascular resistance index was 412, which militates in favour of precapillary PH.
In order to rule out pulmonary disease, chest computed tomography (CT) and pulmonary function test (PFT) were performed. According to chest CT, no structural lung pathology was revealed. According to PFT, moderate restrictive impairment was shown. Also, the contrast-enhanced PA CT was performed, according to which there were no data suggestive of thrombosis (Figure 7).
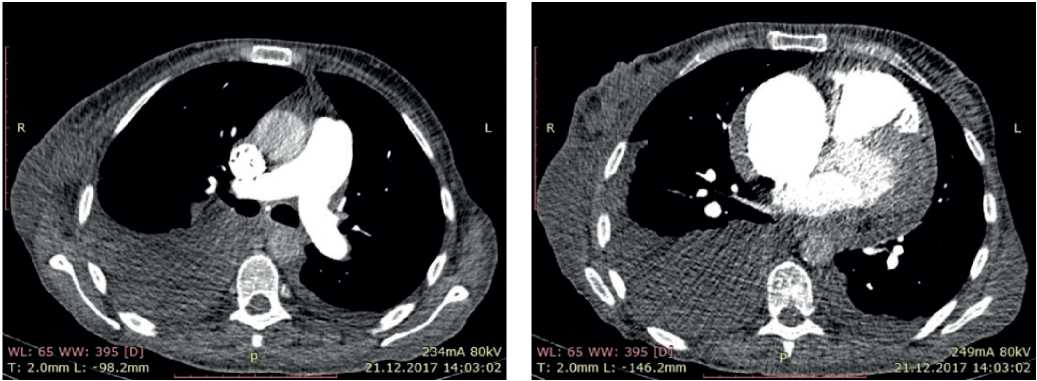
Figure 7. CT of 50-years-old female patient: axillary lymphadenopathy. Hydropericardium. Bilateral hydrothorax.
In addition, PH can most often be caused by congenital heart defects (atrial septal/IVS defects, patent ductus arteriosus, partial anomalous pulmonary venous return), as well as HIV infection and portal hypertension. According to transesophageal echocardiography, contrast-enhanced cardiac CT, abdominal ultrasound, and laboratory tests, these reasons were ruled out and HIV infection was not detected.
The patient’s age at the onset and the presence of concomitant systemic syndromes made it unlikely that PH was hereditary.
Subsequently, the diagnostic search was aimed at identifying pathologies included in the PH group with unclear and/or multiple mechanisms. The presence of lymphadenopathy, articular syndrome, hepatomegaly made it possible to suspect the lymphoproliferative disease.
Morphological and immunohistochemical analysis of biopsy specimens:
Morphology: skin — epidermis with hyperkeratosis, thinned; in the basal layer, the lymphoid infiltrate with a pagetoid growth, pronounced exocytosis, which is represented by medium-sized cells with abnormal nuclear shape and pronounced nuclear folds. Lymph nodes — the lymph node structure is disturbed due to a sharp paracortical expansion, infiltrated by polygonal cells with abnormal nuclear shape and nuclear folds. The pre-existing follicles are small with a thinned mantle zone and are pushed aside by tumour infiltrate. The sinuses are dilated. Inner layer with lipomatosis (Figures 8-10). Conclusion: the immunomorphological picture of Sezary syndrome with lymph node involvement. Diagnosis: Sezary disease — T-cell lymphoma with lymph node involvement.
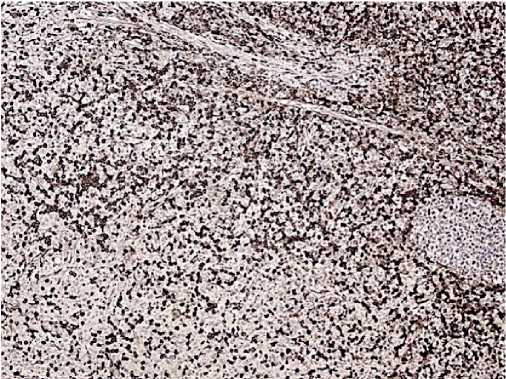
Figure 8. CD4, clone 4B12. Positive reaction is in most tumour cells.
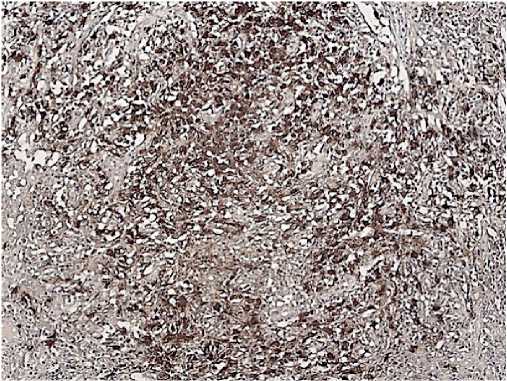
Figure 9. CD8, Clone C8/144B. Positive reaction is in some tumour cells
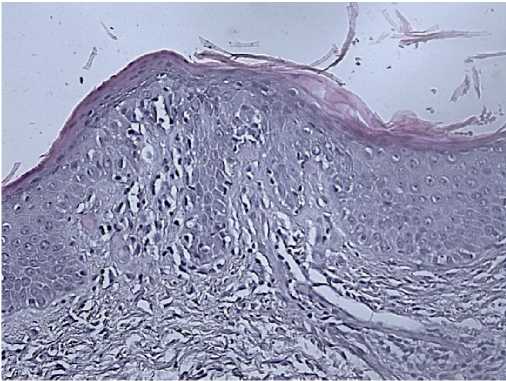
Figure 10. Skin biopsy of 50-years-old female patient.
Final diagnosis: Sezary disease (grade III). Severe PAH associated with lymphoproliferative disease (Sezary disease). Pulmonary heart disease, stage IIB heart failure.
With diuretic and heart rate reduction therapy, exercise tolerance increased and oedema regressed. After 4 years of follow-up, the patient is alive, feels satisfactory, and receives the recommended therapy.
Sezary disease is an extremely rare disease — 3 cases per 1 million population [2][3], which is a type of T-cell lymphoma (peripheral CD4+ T-cells are involved) and is characterized by generalized erythroderma, lymphadenopathy and presence of Sezary cells in the skin, lymph nodes and peripheral blood [3][4].
Early diagnosis of this disease is important in order to initiate adequate therapy, since cases of complete recovery or long-term remission in these patients are rare [5].
According to the literature, cases of sever PH are described in patients with B-cell lymphoma. Such cases are extremely rare in T-cell lymphomas [4][5].
Discussion
The presented case, according to the latest classification, belongs to the fifth group of PH causes.
According to the 2008 WHO classification of lymphomas, mycosis fungoides/Sezary disease refers to primary T-cell lskin lymphomas from mature (peripheral) cells [2]. The disease was first described by the French dermatologist Alibert in 1806. In 1832, he proposed the name, emphasizing the clinical manifestations — fungous tumors. In 1879, the French dermatologist Bazin described in detail the clinical performance of the disease, and since then it bears the name of Alibert-Bazin. The first description of the disease in Russia was made by A.M. Stukovenkov in 1889 [1][5]. In 1938, the French dermatologist Cesari first described a 59-year-old woman with generalized erythroderma, lymphadenopathy, intense itching, and abnormal monocytoid cells in the peripheral blood [1]. Sezary disease accounts for 2-3% of all lymphomas [2]. It is considered as a leukemic type of disease, characterized by chronic erythroderma with diffuse lymphoid dermal infiltration, release of abnormal lymphocytes into the blood, and damage to the bone marrow [4]. Classic Sezary cells have large folded nuclei similar to the brain. They are mainly localized in the upper layers of the dermis and epidermis and have an immunological phenotype of T-helpers (CD2+, CD3+, CD4+, CD5+) [2].
This diagnosis is established by comprehensive assessment of clinical picture, histological and immunophenotypic studies of skin biopsies, and determination of the T-cell receptor gene rearrangement. It should be noted that currently there are no uniform generally accepted diagnostic criteria for mycosis fungoides, and clinical guidelines differ significantly in the amount of recommended diagnostic tests. Clinical examination of the patient remains the fundamental method in diagnosis, because makes it possible to determine the type and stage of this disease, but with the leukemic type of cutaneous T-cell lymphoma, the importance of complete blood count increases [5].
Lung involvement in T-cell lymphomas occurs in about 10% of cases [6]. The main mechanism of LH development in these patients is considered to be the stimulation of vascular endothelial cells by factors associated with platelets, primarily serotonin and growth factor [7]. Other probable reasons are considered to be increased blood viscosity with abnormal immunoglobulin function [8, 9] and tumour microembolism [8].
Conclusion
In the presented case report, a sequential diagnostic algorithm was demonstrated, which made it possible to establish a rare cause of PH. Given the infrequent reference of Sezary disease in cardiology literature, the unification of dissociated syndromes into a single disease is difficult.
The absence of guidelines on PH treatment in patients with lymphoproliferative diseases requires an individual approach to PAH-targeted therapy in each case.