Familial hypercholesterolemia: case series of a rare condition
Introduction
Familial hypercholesterolemia (FH) is the most common autosomal dominant disease caused by mutations in the low-density lipoprotein receptor (LDLR), apolipoprotein B-100 (ApoB), and proprotein convertase subtilisin/kexin type 9 (PCSK9) genes [1][2] leading to an increase in low-density lipoprotein (LDL) level, early atherosclerotic lesions and cardiovascular events at a young age (Figure 1) [3]. The homozygous FH form is a severe but rare hereditary disease, while the heterozygous one is quite common. According to the latest estimates, the prevalence of FH in the world is 1 in 220 people [4], while in Western Siberia it is even higher — 1:108 [5]. As before, <1% of the population in different countries has a verified diagnosis and receives treatment [3]. Although early initiation of therapy can reduce the progression of atherosclerosis, while in most cases the disease is diagnosed after the development of complications: FH leads to myocardial infarction (MI) at the age of <45 years in 20% of cases [6][7]. At the time of FH diagnosis, up to 30% of patients already have a history of coronary artery intervention [8].
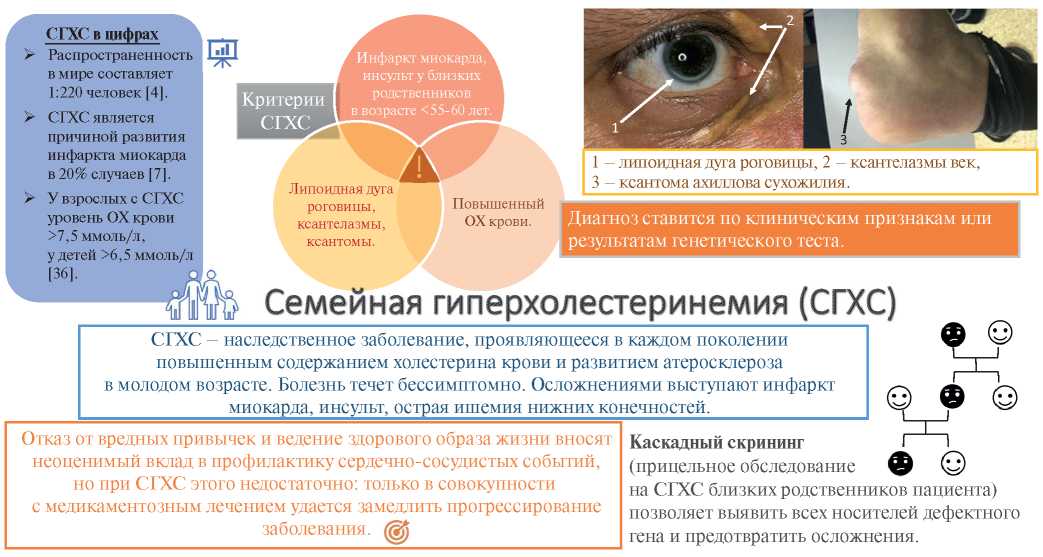
Figure 1. FH — schematic summary.
Abbreviations: TC — total cholesterol, FH — familial hypercholesterolemia.
Unfortunately, even those patients who are aware of their pathology often do not achieve safe cholesterol levels. According to a large European meta-analysis that included studies of 2006-2017 (81 studies, 303534 high- and very high-risk patients), which also examined patients with FH (11 studies, 41594 patients), only 35% received statin therapy, and only 15% (9-22%) reached LDL <2,5 mmol/l [9]. According to the RENESSANS registry (Russia), which included 1208 patients with FH with an average LDL level of 6,6±2,1 mmol/l, the proportion of patients taking lipid-lowering therapy was 72%, while in a high-intensity regimen — 61%. The level of LDL <2,5 mmol/l was reached by 13%, and the level of <1,8 mmol/l was reached by only 6% [10]. Non-achievement of targets is associated both with insufficient adherence to therapy and with a small proportion of patients receiving highintensity therapy with a PCSK9 inhibitor. According to the PLANET registry (Czech Republic and Slovakia) (n=1755), among patients with FH taking PCSK9 inhibitors (n=55), 61,8% achieved the target LDL level [11]. According to the same register, cardiovascular events occurred only in 14% of cases in patients with a target LDL value.
Case reports of FH
Currently, genetic research in order to identify specific mutations responsible for FH has become increasingly available in Russia. Despite the high cost of research, the data obtained from genome sequencing provide useful additional information, evaluating a particular individual in more depth and making it possible to determine the intensity in achieving LDL targets. In addition, knowledge about the type of mutation often makes it possible to make a choice in favor of one or another lipidlowering therapy.
Case report 1. We follow up a mother (born in 1961) and a son (born in 1990) with a confirmed diagnosis of FH. Both have no prior cardiovascular events. The mother showed an increase in total cholesterol (TC) up to 7,3 mmol/l (LDL, 6,2 mmol/l). The son’s maximum TC level during his life was 7,86 mmol/l (LDL, 5,65 mmol/l). Both patients take statins, which significantly decrease LDL values: mother, ~2,3 mmol/l with rosuvastatin therapy; son, 2,49 mmol/l (rosuvastatin of 20 mg). Genetic study revealed the same heterozygous mutation of the ApoB gene (NP_000375.2:p. Arg3527Gln/NC_000002.11:g.21229160C>T) localized in exon 26. This variant is the most common defect found in the ApoB gene, with an overall prevalence of 6-10% among individuals with FH [12]. Various studies have shown that carriers of this variant who develop hypercholesterolemia had a milder phenotype in terms of lipid levels and age at onset of coronary artery disease (CAD) than LDLR mutation carriers [13]. The median age of CAD onset is 50 years. Achievement of LDL levels in the son <2,5 mmol/l (moreover, from the age of 22), i.e. normal for a person without FH, suggests that the age of onset of a possible cardiovascular event will be delayed until at least 60-65 years of age.
It is noteworthy that, in addition to the ApoB mutation, the mother also had a mutation in the PCSK9 gene (NP_777596.2:p.Arg46Leu/ NC_000001.10:g.55505647G>T), which has a protective effect and is absent in the son. This mutation is a widely studied polymorphism [14] that acts as a genetic modifier of LDL levels and response to statins. The p.Arg46Leu polymorphism in PCSK9 is associated with a significant decrease in LDL levels compared with the control group and a lower prevalence of tendon xanthomas [15]. Most importantly, the prevalence of ?1 cardiovascular event in p.Arg46Leu variant carriers was 11% compared to 33% in control group, and this difference was significant (p=0,05).
Thus, a genetic study of this family indicates the need for early statin therapy initiation in ApoB carriers and reveals a potentially better longterm prognosis in a mother who is a carrier of the protective PCSK9 gene.
Case report 2. A 41-year-old man with prior percutaneous coronary interventions (PCI) at the age of 27 and 33 with a total of nine stents implanted. The maximum TC level was 14 mmol/l at the age of 27. In the family history, the early father death from a recurrent MI at the age of 57 (first MI at 27 years of age) draws attention. A genetic study revealed a homozygous ApoB mutation (NP_000375.2:p. Arg3527Gln/NM_000384.2:c.10580G>A/ NC_000002.11:g.21229160C>T). During therapy with 80 mg atorvastatin, it was possible to reduce TC level to 7,88 and LDL to 6,02 mmol/l. Further, a combination with 10 mg ezetimibe and 140 mg evolocumab was added. As can be seen in comparison with example 1, the homozygous type of inheritance determines the severity of atherosclerosis, initially higher levels of TC and LDL, and the need for combined cholesterol-lowering therapy to achieve target LDL values.
Case report 3. A young female patient, born in 2000, with a positive family history of cardiovascular disease (paternal grandfather had sudden cardiac death at 44 years old; father had confirmed coronary atherosclerosis without indications for revascularization at 41 years old) and an increase in TC to 10,26 mmol/l and LDL to 8,5 mmol/l. The genetic analysis showed a null mutation in LDLR gene (NM_000527.4:c.2389+5G>C/ NC_000019.9:g.11238766G>C). This mutation was previously found only in 2 cases in Russian patients in the Health in Code library (La Coru?a, Spain). However, mutation in the LDLR gene causes an extremely high risk of early atherosclerosis and, possibly, a suboptimal effect of standard lipidlowering therapy. So, in the patient’s father (most likely having the same mutation), high-intensity statin therapy (rosuvastatin 40 mg) reduced the TC level to 7,24 mmol/l and LDL to 4,95 mmol/l (initial documented values of TC ~12 mmol/l). The patient herself does not take cholesterol-lowering drugs.
Case report 4. We follow up a family, 6 members of which have definite FH according to the Dutch Lipid Clinic Network (DLCN) criteria (Figure 2). The first person was a 34-year-old young man (proband), in whom CAD manifested with myocardial infarction, followed by PCI. In lipid profile, an increase in TC to 8,65 mmol/l and LDL to 6,6 mmol/l was revealed. When taking a detailed history, it turned out that the patient’s brother died due to myocardial infarction at the age of 32; the mother has a high class angina at the age of 55; a maternal uncle had PCI at age 43; maternal grandfather died at 42 from a myocardial infarction. All of these family members had a documented TC level >7,5 mmol/L. In addition, the proband’s daughter has an TC level of 9 mmol/l. The proband’s mother underwent coronary artery bypass grafting 1 month after the FH detection due to three-vessel disease. This example demonstrates the importance of cascade screening and the possibility of applying clinical criteria for the diagnosis of FH and identification of risk groups for early development of cardiovascular events.
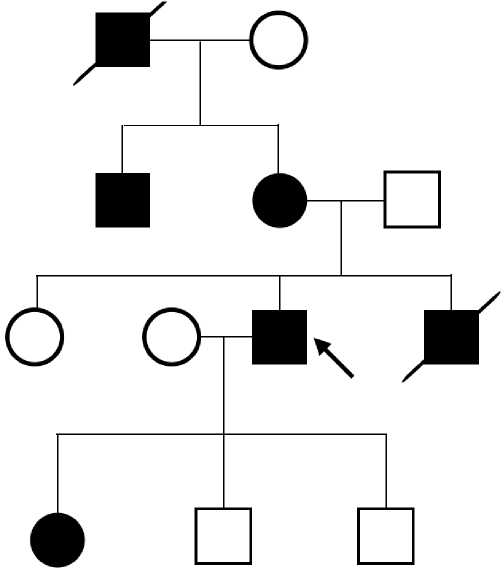
Figure 2. Pedigree of a patient with severe hypercholesterolemia.
In preparing this article, the patient of case 2 died from a recurrent cardiovascular event. Follow-up of the rest of patients continues.
Discussion
Screening
All over the world there is an organized system for the early diagnosis of hereditary diseases, such as phenylketonuria, congenital hypothyroidism, etc., the purpose of which is to prevent long-term and irreversible consequences of metabolic disorders. Atherosclerosis, which develops as a result of improper lipid metabolism, progresses imperceptibly and complicates in the form of myocardial infarction and strokes. This is a burden not only for a single family, but also for the state as a whole, especially when it comes to the working-age population. The total economic damage from hypercholesterolemia in Russia is estimated at 1295 trillion rubles per year, including direct costs (emergency care, disability benefits) and indirect losses (temporary disability; loss of gross domestic product due to disability; loss of gross domestic product associated with premature death in economically active age) [16]. The life expectancy of men with heterozygous FH in our country is 53 years, women — 62 years [17].
Every year in Russia more patients with severe lipid metabolism disorders are included in the registries, new lipid centers are opened, but the system for identifying and monitoring patients with FH still needs to be improved. There are many reasons for this: lack of awareness of doctors, low medical adherence of patients, and lack of lipid centers and low availability of genetic diagnostic methods in the regions. Certain difficulties arise with the choice of a screening method for identifying FH patients. Although hypercholesterolemia is an independent risk factor for atherosclerosis, an isolated increase in cholesterol does not predict FH with a high accuracy. In addition, recent studies have not demonstrated a high FH incidence in the population of patients with severe hypercholesterolemia (0,3-1,7%) [18][19], and therefore the cost-effectiveness of such screening. Wald DS, et al. (2007) proposed to measure TC level in all children aged 1-9 years, because it turned out that it is at this age that the greatest difference in TC levels of healthy children and carriers of mutations is determined (it is possible to diagnose FH in 88% with 0,1% of false positive results) [20]. The authors argue that if this segment of the population is subjected to genetic testing followed by cascade screening, then in 30 years the majority of carriers of defective genes will be known, which makes it possible to stop continuous screening and only monitor the affected families. Cascade screening included examination of the patient’s first-, second-, and sometimes third-degree relatives for an already identified mutation. This method allows to detect FH before the development of complications for primary prevention. Later, the same authors examined 10095 children aged 1-2 years during scheduled visits to the polyclinic; as a result, FH was detected in 40 children (0,4%) and 40 parents [21]. According to the study, neither parent had previously received lipid-lowering therapy. It should be noted that only severe hypercholesterolemia (>95 percentile) was considered by the authors as screening for FH.
Another method is risk group search and targeted screening. Patients with prior acute coronary syndrome (ACS) aged less than 55 and 60 years in men and women, respectively, represent a promising cohort for such screening. According to the metaanalysis with 22 studies including 31436 cases, FH prevalence increases with decreasing age of patients. Among patients <60 years of age, the prevalence was 7,3%, and when narrowing the sample to age <45 years, the prevalence of probable/definite FH increased to 13,7% [22]. In the case of ACS, all patients with FH are at very high risk, however, given their young age, the absence of known comorbidities and prior cardiovascular events, when deciding on management tactics, such patients may be erroneously classified as low risk, which will lead to prolongation of intravascular intervention and worse prognosis in case of MI [23].
Diagnostics
Although the gold standard for hereditary disease verification is the genetic method, it is not always available. For this reason, several variants of clinical criteria for FH diagnosis have been developed. Among them, the most famous are British (SB — Simon Broome Registry) [24], Dutch (DLCN — Dutch Lipid Clinic Network) [25], American (MEDPED — Make Early Diagnosis to Prevent Early Deaths) [26] and Japanese (JAS — Japanese Atherosclerotic Society) [27]. All scales evaluate the patient’s family history and LDL level. In addition, the Japanese criteria assess the Achilles tendon thickness, while SB and DLCN assess the presence of a genetic mutation. DLCN also take into account a patient’s history of cardiovascular events and are the most detailed in assessing the FH likelihood. The direct comparison of clinical criteria and genetic testing for FH diagnosis in 103 patients ?65 years of age with ACS, the following results were obtained: the prevalence of confirmed FH was 8,7%; DLCN and SB criteria did not diagnose FH in 4 and 3 cases, respectively [28]. Low sensitivity may be related to the widespread use of statins among the population and artificially reduced LDL values. The availability of plastic surgery allows many patients to dispose of xanthomas and xanthelasmas [29], which increases clinical suspicion. Thus, the presented criteria occupy an important place in FH diagnosis, but cannot completely replace the genetic method. The need for genetic testing arises in patients with definite FH and no response to high-dose lipid-lowering therapy. Some mutations, such as homozygous null mutations in the LDLR gene, require lipid apheresis or liver transplantation because patients are insensitive to statins and PCSK9 inhibitors [30-32]. There may be a similarity of clinical manifestations of FH and other genetic dyslipidemias, such as familial hypertriglyceridemia, sitosterolemia, dysbetalipoproteinemia, etc. [32][33]. These patients also often require specific therapy other than the standard treatment for FH.
Masana L, et al. (2019) propose to revise FH classification, considering it as a syndrome, i.e. a combination of hypercholesterolemia, family history and high cardiovascular risk due to various causes. The FH syndrome includes both clinical and genetic variants as follows: heterozygous FH; homozygous FH; polygenic FH; FH combined with hypertriglyceridemia [34]. Carriers of mutations in the genes of apolipoprotein E, Signal transducing adaptor protein (STAP1), and patatin-like phospholipasecontaining domain 5 (PNPLA5) should also be classified as heterozygous FH [2]. In addition to homozygotes for the LDLR, ApoB or PCSK9 genes, it is also proposed to refer to the homozygous form as compound heterozygotes (both alleles of one gene have different mutations), combined heterozygotes (two genes have different mutations) and autosomal recessive hypercholesterolemia with low density lipoprotein receptor adapter protein 1 (LDLRAP1) mutation. The authors suggest diagnosing polygenic FH if the known criteria are met and there are no mutations of the above-mentioned genes [34]. From 10 to 50% of patients can be attributed to polygenic FH according to various studies [3][35]. Although the risk of such patients may be lower than those with known monogenic mutations, they also need active prevention and monitoring from a young age.
Prevention
According to the 2019 European Society of Cardiology guidelines for dyslipidemia, patients with FH and without atherosclerotic diseases are at high risk of cardiovascular complications with a target LDL <1,8 mmol/l, and if they are present, while in case of their presence — at very high risk with target LDL <1,4 mmol/l or a decrease in LDL by ?50% [6]. According to the Russian Society of Cardiology guidelines for the management of FH patients, the target LDL level in adult patients with FH should be <2,5 mmol/l; with cardiovascular diseases — <1,5 mmol/l; for children under 10 years of age — <4 mmol/l, and after reaching 10 years — <3,5 mmol/l [36]. It is recommended to start lipid-lowering therapy at the age of 8-10 years if non-drug methods are ineffective within 3-6 months [6][36][37]. The results of the following study confirm the need for primary prevention in children. The authors took a cohort of 214 FH patients aged 31,7±3,2 years (genetically confirmed, 98%) who participated in a 2-year pravastatin study as children. The researchers measured LDL levels and assessed the history of cardiovascular events/death for 18 (15- 21) years. In addition, 156 parents with FH were also studied for adverse events. The following results were obtained: 79% of patients received lipid-lowering therapy; among those who stopped taking, only four cases had side effects; no cases of rhabdomyolysis or other serious conditions were noted; among those who continued therapy, no increase in liver enzymes or creatine phosphokinase was obtained. The control group consisted of siblings of patients (n=95) who did not suffer from FH [38]. Thus, the safety of longterm statin therapy in children has been demonstrated. The LDL level decreased by 32% compared to the original. At the same time, MI, angina, peripheral arterial disease, stroke, myocardial revascularization or cardiovascular death occurred in 1% and 0%, respectively, in patients, in 26% and 7%, respectively, in their parents who were unavailable to therapy in early age [38].
Heredity plays a significant, but not decisive role. The CASCADE-FH registry, which includes >1900 patients with FH, has shown that risk factors such as age, male sex, hypertension, diabetes, obesity, and smoking lead to an increased incidence of cardiovascular events in FH individuals [39]. Similar data were obtained by Akioyamen LE, et al. (2019) in a meta-analysis [40].
Conclusion
The life expectancy of a patient with FH is directly dependent on the effectiveness of both primary and secondary prevention of cardiovascular events; which are possible under certain conditions:
- screening of patients with early cardiovascular events;
- routing of patients with suspected FH to lipid centers;
- availability of genetic analysis; the use of clinical criteria in case of its impossibility;
- raising physicians’ awareness of FH and conducting cascade screening for more effective prevention.
Registries allow not only to structure the information already available, but also to obtain information on the influence of additional risk factors on disease course, the effectiveness of therapy and prognosis for life in patients with already known genetic mutations. Future research will help determine the best method for early FH screening, and the annual increase in the disease prevalence will indicate in favor of better diagnosis, and therefore control of the situation. The authors did not regard drug prevention of cardiovascular events. However, clinical studies in this area will also significantly improve the prognosis of FH patients in the future.
Patients in the presented case reports have given their consent to the publication of information without disclosing names.
Relationships and Activities: none.