Contribution of electrocardiography to the diagnosis of cardiomyopathies and athletic heart syndrome
Electrocardiography (ECG) is the most accessible and reproducible method for cardiac assessment. For a long time, in nonischemic myocardial diseases, abnormalities in the ECG were considered nonspecific. Recent studies with modern technologies, such as magnetic resonance imaging (MRI) and genetic testing, have made significant progress in understanding pathological processes in the myocardium and defining specific ECG abnormalities for some of them. In cardiomyopathies, ECG abnormalities observed in coronary artery disease and hypertension (HTN) have a different origin and are due to microcirculation disorders, interstitial fibrosis, disorganized cardiomyocytes or their fibro-fatty replacement, as well as asymmetric hypertrophy, changing the QRS axis. The correct interpretation of ECG abnormalities often allows one to promptly assume the true nature of the disease. In some cases, ECG abnormalities are the only phenotypic manifestation of inherited heart disease [1], which makes the method indispensable for family screening.
In athletes, more often in dynamic sports (cycling, football, running), long-term intense exercise leads to structural and electrical adaptive changes, which are commonly called the “athletic heart syndrome” [2]. These changes are benign in most cases, but sometimes they can lead to cardiomyopathies, which are the leading cause of sudden cardiac death (SCD) in young athletes [3]. The correct interpretation of the ECG in athletes, on the one hand, can help to timely diagnose a fatal disease, and on the other hand, to avoid false disqualification [3][4].
Arrhythmogenic cardiomyopathy
Arrhythmogenic cardiomyopathy (ACM), previously defined only as a arrhythmogenic right ventricular (RV) dysplasia, is a genetic disease of the myocardium of RV and/or left ventricle (LV). The distinctive phenotypic feature of ACM is the scarring in the form of fibrous or fibro-fatty replacement of cardiomyocytes which serve as a substrate for global and/or local myocardial dysfunction and predispose to fatal ventricular arrhythmias [5]. The diagnosis of ACM is collective and is based on a combination of morphological, functional, and structural myocardial changes, revealed by echocardiography, MRI, biopsy, resting ECG, and 24-hour ambulatory ECG monitoring. Analysis of family history and genetic testing also plays an important role in the diagnosis of ACM.
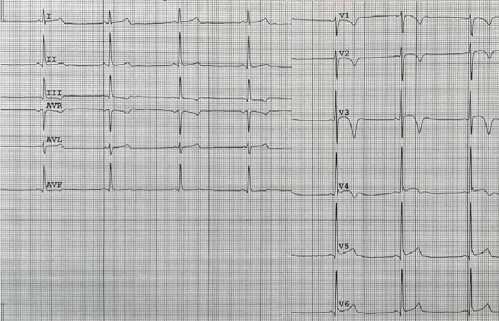
On the ECG of ACM patients, there are criteria specific for the predominant involvement of the RV or LV, which are subdivided into major and minor [5]. Thus, inverted T-waves in the right-sided chest leads (V1-V3) in adults without complete right bundle branch block (RBBB) is a major criterion for right-dominant ACM. The spread of inverted T-waves to V4-V6 indicates significant RV dilatation and dysfunction (Figure 1). In the case of complete RBBB, such inversions become less specific and refer to the minor criteria of right-dominant ACM. Also, diseases that may resemble ACM should always be ruled out, such as heart displacement due to pericardiotomy or chest wall deformity, RV volume or pressure overload, cardiac sarcoidosis, and myocarditis [6].
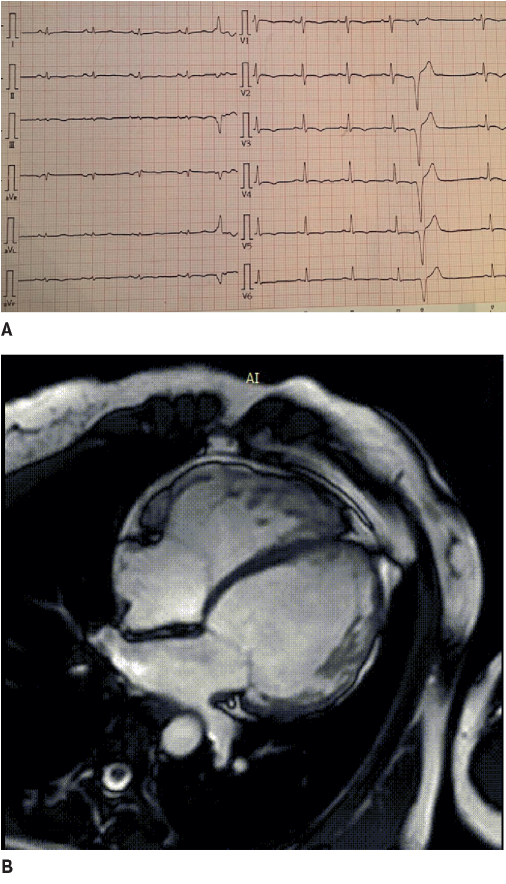
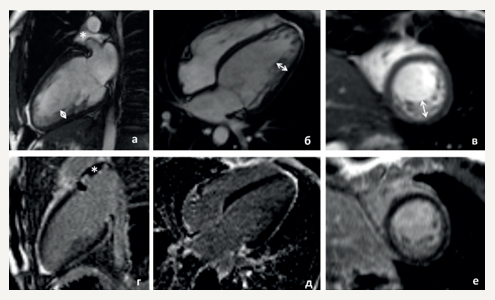
Figure 1. The 42-year-old female patient with biventricular ACM and dominated LV involvement. A. ECG: low QRS voltage in limb leads and inverted T wave in V1-V6; frequent premature ventricular contractions with a configuration of complete LBBB and the superior axis. B. MRI: biventricular dilatation, LV NCM.
Epsilon wave, previously referred to major ACM criteria, is a reproducible low-amplitude signal between the QRS end and T-wave beginning. Over the past ten years, the diagnostic value of this criterion has been questioned due to its various interpretation [7], and in the 2020 updated Padua criteria [5], the epsilon wave is attributed to the minor criterion of right-dominant ACM, as well as terminal activation duration >55 ms, measured from the nadir of the S wave to the end of the QRS, including R’, in V1, V2, or V3 (in the absence of complete RBBB).
Low QRS voltage in limb leads (<5 mm) may indicate LV involvement in ACM (Figure 1). The sensitivity of this criterion is low (<30%); therefore, it is considered minor for the left-dominant ACM in the absence of obesity, emphysema, or pericardial effusion [5]. Also, minor criteria for left-dominant ACM include inverted T waves in left precordial leads (in the absence of complete left bundle branch block (LBBB)) [5]. The isolated left-dominant ACM is phenotypically indistinguishable from dilated cardiomyopathy (DCM) and is often confirmed only by genetic testing.
Registration of late potentials using a signal-averaged ECG has not found wide application in practice and is no longer used for the diagnosis of ACM.
Ventricular arrhythmia with a configuration of complete LBBB and the inferior axis, which indicates its origin from the RV outflow tract is a minor criterion, and without the lower axis, it is a major criterion for right-dominant ACM [5] (Figure 1). Ventricular arrhythmia with a configuration of complete RBBB is a minor criterion for left-dominant ACM [5].
Myocardial changes similar to ACM, such as significant RV enlargement, borderline decrease in the RV ejection fraction (EF), and ventricular arrhythmias, can be induced by regular exercises in healthy individuals [8][9]. The physiological RV changes on the ECG include voltage criteria for RV hypertrophy, isolated complete RBBB, and right axis deviation Highly sensitive to ACM inversed T-wave, in the right-sided chest leads loses its specificity in athletes due to a fairly high prevalence (up to 4% in V1-V3 and 10% in V1-V2) [2]. According to our unpublished data based on the analysis of 619 ECG records of athletes, inversed T-waves in V1-V3 without any significant structural changes in the heart occur in 1,9% of cases (Figure 2). Nevertheless, today in white athletes, any inversed T-waves in two contiguous leads, including in V1-V3, is regarded as pathological and requires in-depth examination and follow-up. In black athletes, such inversions, especially with ST- segment elevation and a J-point >1 mm, are referred to as benign [10]. In addition to inversed T-waves, ACM can be suspected in athletes when recording ventricular arrhythmias and epsilon waves.

Figure 2. A. The 24-year-old male athlete, pentathlon, asymptomatic. Echocardiography, cardiac MRI, 24-hour ECG monitoring did not reveal any pathology. ECG: inverted T-wave in V1-V3 with preceding ST elevation at J-point >1 mm. B. The 35-year-old female athlete, cycling, asymptomatic, without structural heart disease. ECG: inverted T wave in V1-V3, isolated increase in QRS voltage.
LV hypertrophy
Hypertrophic cardiomyopathy (HCM) is the most common inherited heart disease. In 60% of patients, HCM is caused by sarcomere gene mutations. In 5-10%, HCM is simulated by rare storage diseases (Fabry, Danon, PRKAG2 cardiomyopathy), infiltrative diseases (amyloidosis, sarcoidosis), mitochondrial, neuromuscular diseases (Friedreich’s ataxia), malformations (Noonan syndrome), and endocrine cardiomyopathies. In the remaining 30% of patients, the cause of HCM has not yet been clarified [11]. Unlike ACM, the criteria for the HCM diagnosis are only morphological: LV myocardial thickening in adults >15 mm (>13 mm in the presence of a relative with HCM), which cannot be explained by other conditions leading to LV overload (HTN, aortic valve stenosis) [12].
ECG abnormalities, mainly inversed T-waves or deep narrow (“dagger-like”) Q waves with a positive T wave in the inferior and lateral leads, are recorded in more than 90% of patients with sarcomeric HCM [13]. “Giant” (>10 mm) symmetric T waves, usually present in all chest leads, indicate severe hypertrophy of the LV apex [14] (Figure 3). Pseudo-infarct QS complexes in the chest leads and complete bundle branch blocks occur infrequently in HCM and mainly after surgical reduction of the interventricular septum or in severe transmural fibrosis [13]. Such changes are more typical for infiltrative diseases [15][16] (Figure 4).
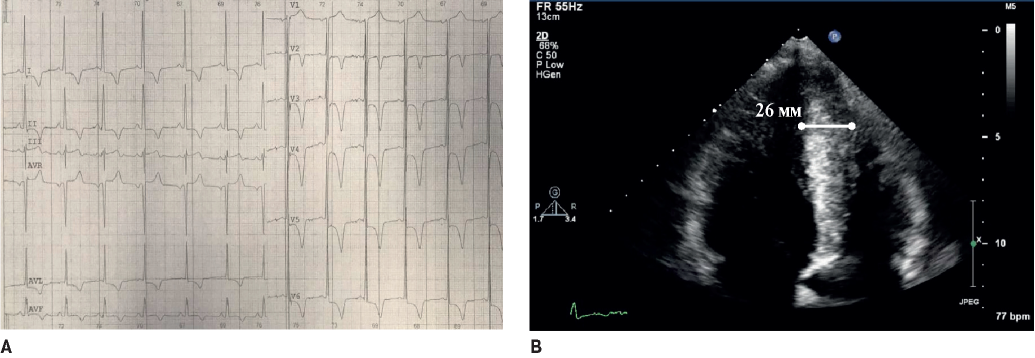
Figure 3. The 34-year-old male patient with the apical familial HCM. A. ECG: “giant” inverted T-waves in V2-V6, LV hypertrophy voltage signs. B. Echocardiography: hypertrophy of LV apex with ace-of-spades sign.
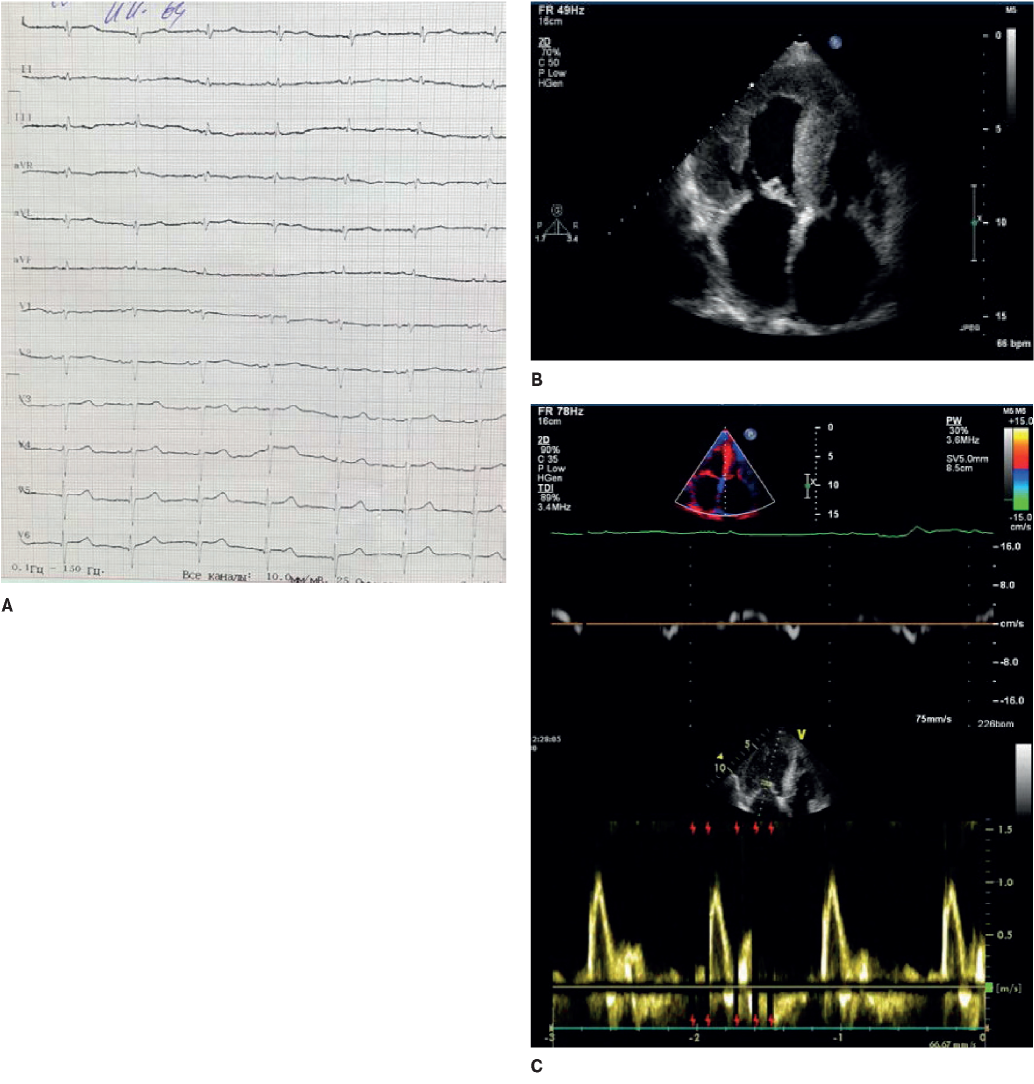
Figure 4. The 64-year-old female patient with familial transthyretin amyloidosis of the heart. A. ECG: low QRS voltage in limb leads and “normal”, not corresponding to the hypertrophy severity on echocardiography, in the chest leads; R regression in V1-V4. B. Echocardiography: severe LV hypertrophy, atrial dilatation. C. Doppler echocardiography: a restrictive LV diastolic dysfunction.
The combination of visually severe LV hypertrophy with conduction abnormalities on the ECG is always suspicious for HCM phenocopies. Thus, a shortened PQ interval should be suggestive of storage [17] or mitochondrial diseases [1], while an atrioventricular conduction delay suggests amyloidosis [15], sarcoidosis [16], or end-stage storage and mitochondrial diseases [1][13][17].
In many patients with HCM, voltage criteria for LV hypertrophy are recorded, and only in 2% they are not accompanied by impaired repolarization [18]. If the voltage is very high, then it is worth suspecting the storage disease [13]. If, on the contrary, the QRS voltage is reduced or normal with severe hypertrophy on ECG, then amyloidosis should be suspected [15] (Figure 4).
Every eighth patient with HCM has elongated QT interval >480 ms, and every second patient has >450 ms, which is associated with the risk of SCD and is an additional argument for implantation of a cardioverter-defibrillator [13][19].
About 5-10% of patients with HCM phenotype have either a normal ECG or an isolated increase in QRS voltage. In such patients, the disease debuts later, the symptoms are less pronounced and the prognosis is better [13][20].
Physiological hypertrophy in athletes does not exceed 14 mm in men [21] and 12 mm in women [22]; nevertheless, it is always suspicious of the HCM onset. One of the most characteristic ECG signs of an athlete’s heart is a pronounced increase in QRS voltage, which is often mistakenly considered as LV hypertrophy. Unlike pathological hypertrophy, there are no concomitant repolarization abnormalities on an athlete’s ECG, therefore, a moderate LV wall thickening on echocardiography in combination with an isolated increase in QRS voltage indicates physiological myocardial remodeling. Repolarization disorders in the form of inverted T wave >1 mm in more than 2 contiguous inferior (II and aVF) and, especially, lateral (I, aVL, V5 or V6) leads indicate a possible HCM [4].
Inverted T wave in the inferior and lateral leads are recorded on the ECG of athletes without structural cardiac changes. Isolated T wave inversions in the inferior leads are found in 2% of white and 6% of healthy black athletes [13], which is much more frequent than inherited heart diseases. According to our unpublished data with 1435 ECGs of athletes from various sports, isolated T wave inversions in the inferior leads are found in 1% of cases. Inverted T waves in the lateral leads are considered the most unfavorable, since they may be the first sign of cardiomyopathy [23]. Abnormalities suspicious of HCM in athletes also include pathological Q waves (>0,25 from the R wave or >40 ms), ST depression >0,5 mm in >2 contiguous leads, complete LBBB, non-specific prolonged QRS >140 ms, and frequent premature ventricular contractions [4].
LV systolic dysfunction
Dilated cardiomyopathy (DCM) is a syndrome characterized by systolic dysfunction and LV dilatation, which cannot be explained by coronary artery disease or conditions leading to LV overload (HTN, valvular and congenital heart disease). LV systolic dysfunction (LVEF <45%) without dilatation since 2016 has been classified as hypokinetic non-dilated cardiomyopathy [24].
DCM is the most etiologically heterogeneous cardiomyopathy. About 40% of DCM cases are inherited [25], which can manifest as isolated heart disease, in combination with conduction defects and noncompacted myocardium (NCM), or within the systemic muscle diseases. Among the latter, the most common DCM phenotype occurs in muscular dystrophies (Duchenne and Becker), limb-girdle muscular dystrophies (LGMD), and Emery-Dreifuss muscular dystrophy (EDMD) [26]. Familial DCM occurs due to mutations in the genes of sarcomere (titin), cytoskeleton (dystrophin, desmin), cell membranes (lamin, ion channels), and organelles [25]. Acquired DCMs develop due to infections, autoimmune diseases, toxic (alcohol, cocaine) or medication (chemotherapy) myocardial damage, micronutrient deficiencies, endocrine and metabolic diseases, and pregnancy [24]. Separately, authors distinguish tachycardia-induced cardiomyopathy — a potentially reversible decrease in LV systolic function, which develops with permanent atrial or ventricular tachyarrhythmia [27]. There is evidence that patients with non-familial DCM also have a genetic substrate of the disease [25][28][29][30].
Table 1
Specific signs of cardiomyopathies on resting ECG
| Phenotype | ECG abnormalities Suggested diagnosis | |
|---|---|---|
| Dilatation/impaired RV contractility | inverted T in V1-V3 | major criterion of right-dominant ACM |
| inverted T in V1-V4(V6) | significant RV involvement | |
| ?-wave in V1-V2 | minor criterion of right-dominant ACM | |
| TAD >55 ms in V1-V3 | minor criterion of right-dominant ACM | |
| ? QRS in limb leads | biventricular ACM | |
| inverted T inV4-V6/I, aVL | biventricular ACM | |
| LV hypertrophy | shortened PQ | Fabry, Danon, Pompe, PRKAG2, mitochondrial diseases |
| AV blocks | amyloidosis, end-stage Fabry, Danon, acute myocarditis | |
| ?? QRS voltage | Danon, Pompe | |
| ? or ’normal’ QRS voltage | amyloidosis | |
| right QRS axis deviation | Noonan syndrome | |
| LV systolic dysfunction | > Р/atrial standstill | type 1 and 2 EDMD |
| sinus bradycardia | laminopathy | |
| shortened PQ | DMD | |
| AV blocks | sarcoidosis, laminopathy, EDMD, myotonic dystrophy, desminopathy, Chagas disease, diphtheria, Lyme disease | |
| Q/QS in inferolateral leads | DMD, BMD, sarcoidosis, LGMD | |
| ? QRS | Left-dominant ACM | |
| Complete RBBB | DMD, Chagas disease (+ left anterior fascicular block) | |
| inverted T in V1-V6 | Left-dominant and biventricular ACM | |
| Hypertrabeculation of LV | Complete LBBB | NCM |
| pathological Q waves | ||
| inverted T | ||
Abbreviations: ACM — arrhythmogenic cardiomyopathy, LV — left ventricle, LBB — left bundle branch, EDMD — Emery-Dreifuss muscular dystrophy, NCM — noncompacted myocardium, RV — right ventricle, LGMD — limb-girdle muscular dystrophies, BMD — Becker muscular dystrophy, DMD — Duchenne muscular dystrophy, TAD — terminal activation delay.

Чтобы читать статью войдите с логином и паролем от scardio.ru
Keywords
For citation
Chumakova O.S., Isaeva M.Yu., Koroleva O.S., Zateyshchikov D.A. Contribution of electrocardiography to the diagnosis of cardiomyopathies and athletic heart syndrome. Russian Journal of Cardiology. 2020;25(3S):4023. https://doi.org/10.15829/1560-4071-2020-4023
Electrocardiography (ECG) is the most accessible and reproducible method for cardiac assessment. For a long time, in nonischemic myocardial diseases, abnormalities in the ECG were considered nonspecific. Recent studies with modern technologies, such as magnetic resonance imaging (MRI) and genetic testing, have made significant progress in understanding pathological processes in the myocardium and defining specific ECG abnormalities for some of them. In cardiomyopathies, ECG abnormalities observed in coronary artery disease and hypertension (HTN) have a different origin and are due to microcirculation disorders, interstitial fibrosis, disorganized cardiomyocytes or their fibro-fatty replacement, as well as asymmetric hypertrophy, changing the QRS axis. The correct interpretation of ECG abnormalities often allows one to promptly assume the true nature of the disease. In some cases, ECG abnormalities are the only phenotypic manifestation of inherited heart disease [1], which makes the method indispensable for family screening.
In athletes, more often in dynamic sports (cycling, football, running), long-term intense exercise leads to structural and electrical adaptive changes, which are commonly called the “athletic heart syndrome” [2]. These changes are benign in most cases, but sometimes they can lead to cardiomyopathies, which are the leading cause of sudden cardiac death (SCD) in young athletes [3]. The correct interpretation of the ECG in athletes, on the one hand, can help to timely diagnose a fatal disease, and on the other hand, to avoid false disqualification [3][4].
Arrhythmogenic cardiomyopathy
Arrhythmogenic cardiomyopathy (ACM), previously defined only as a arrhythmogenic right ventricular (RV) dysplasia, is a genetic disease of the myocardium of RV and/or left ventricle (LV). The distinctive phenotypic feature of ACM is the scarring in the form of fibrous or fibro-fatty replacement of cardiomyocytes which serve as a substrate for global and/or local myocardial dysfunction and predispose to fatal ventricular arrhythmias [5]. The diagnosis of ACM is collective and is based on a combination of morphological, functional, and structural myocardial changes, revealed by echocardiography, MRI, biopsy, resting ECG, and 24-hour ambulatory ECG monitoring. Analysis of family history and genetic testing also plays an important role in the diagnosis of ACM.
On the ECG of ACM patients, there are criteria specific for the predominant involvement of the RV or LV, which are subdivided into major and minor [5]. Thus, inverted T-waves in the right-sided chest leads (V1-V3) in adults without complete right bundle branch block (RBBB) is a major criterion for right-dominant ACM. The spread of inverted T-waves to V4-V6 indicates significant RV dilatation and dysfunction (Figure 1). In the case of complete RBBB, such inversions become less specific and refer to the minor criteria of right-dominant ACM. Also, diseases that may resemble ACM should always be ruled out, such as heart displacement due to pericardiotomy or chest wall deformity, RV volume or pressure overload, cardiac sarcoidosis, and myocarditis [6].
Figure 1. The 42-year-old female patient with biventricular ACM and dominated LV involvement. A. ECG: low QRS voltage in limb leads and inverted T wave in V1-V6; frequent premature ventricular contractions with a configuration of complete LBBB and the superior axis. B. MRI: biventricular dilatation, LV NCM.
Epsilon wave, previously referred to major ACM criteria, is a reproducible low-amplitude signal between the QRS end and T-wave beginning. Over the past ten years, the diagnostic value of this criterion has been questioned due to its various interpretation [7], and in the 2020 updated Padua criteria [5], the epsilon wave is attributed to the minor criterion of right-dominant ACM, as well as terminal activation duration >55 ms, measured from the nadir of the S wave to the end of the QRS, including R’, in V1, V2, or V3 (in the absence of complete RBBB).
Low QRS voltage in limb leads (<5 mm) may indicate LV involvement in ACM (Figure 1). The sensitivity of this criterion is low (<30%); therefore, it is considered minor for the left-dominant ACM in the absence of obesity, emphysema, or pericardial effusion [5]. Also, minor criteria for left-dominant ACM include inverted T waves in left precordial leads (in the absence of complete left bundle branch block (LBBB)) [5]. The isolated left-dominant ACM is phenotypically indistinguishable from dilated cardiomyopathy (DCM) and is often confirmed only by genetic testing.
Registration of late potentials using a signal-averaged ECG has not found wide application in practice and is no longer used for the diagnosis of ACM.
Ventricular arrhythmia with a configuration of complete LBBB and the inferior axis, which indicates its origin from the RV outflow tract is a minor criterion, and without the lower axis, it is a major criterion for right-dominant ACM [5] (Figure 1). Ventricular arrhythmia with a configuration of complete RBBB is a minor criterion for left-dominant ACM [5].
Myocardial changes similar to ACM, such as significant RV enlargement, borderline decrease in the RV ejection fraction (EF), and ventricular arrhythmias, can be induced by regular exercises in healthy individuals [8][9]. The physiological RV changes on the ECG include voltage criteria for RV hypertrophy, isolated complete RBBB, and right axis deviation Highly sensitive to ACM inversed T-wave, in the right-sided chest leads loses its specificity in athletes due to a fairly high prevalence (up to 4% in V1-V3 and 10% in V1-V2) [2]. According to our unpublished data based on the analysis of 619 ECG records of athletes, inversed T-waves in V1-V3 without any significant structural changes in the heart occur in 1,9% of cases (Figure 2). Nevertheless, today in white athletes, any inversed T-waves in two contiguous leads, including in V1-V3, is regarded as pathological and requires in-depth examination and follow-up. In black athletes, such inversions, especially with ST- segment elevation and a J-point >1 mm, are referred to as benign [10]. In addition to inversed T-waves, ACM can be suspected in athletes when recording ventricular arrhythmias and epsilon waves.
Figure 2. A. The 24-year-old male athlete, pentathlon, asymptomatic. Echocardiography, cardiac MRI, 24-hour ECG monitoring did not reveal any pathology. ECG: inverted T-wave in V1-V3 with preceding ST elevation at J-point >1 mm. B. The 35-year-old female athlete, cycling, asymptomatic, without structural heart disease. ECG: inverted T wave in V1-V3, isolated increase in QRS voltage.
LV hypertrophy
Hypertrophic cardiomyopathy (HCM) is the most common inherited heart disease. In 60% of patients, HCM is caused by sarcomere gene mutations. In 5-10%, HCM is simulated by rare storage diseases (Fabry, Danon, PRKAG2 cardiomyopathy), infiltrative diseases (amyloidosis, sarcoidosis), mitochondrial, neuromuscular diseases (Friedreich’s ataxia), malformations (Noonan syndrome), and endocrine cardiomyopathies. In the remaining 30% of patients, the cause of HCM has not yet been clarified [11]. Unlike ACM, the criteria for the HCM diagnosis are only morphological: LV myocardial thickening in adults >15 mm (>13 mm in the presence of a relative with HCM), which cannot be explained by other conditions leading to LV overload (HTN, aortic valve stenosis) [12].
ECG abnormalities, mainly inversed T-waves or deep narrow (“dagger-like”) Q waves with a positive T wave in the inferior and lateral leads, are recorded in more than 90% of patients with sarcomeric HCM [13]. “Giant” (>10 mm) symmetric T waves, usually present in all chest leads, indicate severe hypertrophy of the LV apex [14] (Figure 3). Pseudo-infarct QS complexes in the chest leads and complete bundle branch blocks occur infrequently in HCM and mainly after surgical reduction of the interventricular septum or in severe transmural fibrosis [13]. Such changes are more typical for infiltrative diseases [15][16] (Figure 4).
Figure 3. The 34-year-old male patient with the apical familial HCM. A. ECG: “giant” inverted T-waves in V2-V6, LV hypertrophy voltage signs. B. Echocardiography: hypertrophy of LV apex with ace-of-spades sign.
Figure 4. The 64-year-old female patient with familial transthyretin amyloidosis of the heart. A. ECG: low QRS voltage in limb leads and “normal”, not corresponding to the hypertrophy severity on echocardiography, in the chest leads; R regression in V1-V4. B. Echocardiography: severe LV hypertrophy, atrial dilatation. C. Doppler echocardiography: a restrictive LV diastolic dysfunction.
The combination of visually severe LV hypertrophy with conduction abnormalities on the ECG is always suspicious for HCM phenocopies. Thus, a shortened PQ interval should be suggestive of storage [17] or mitochondrial diseases [1], while an atrioventricular conduction delay suggests amyloidosis [15], sarcoidosis [16], or end-stage storage and mitochondrial diseases [1][13][17].
In many patients with HCM, voltage criteria for LV hypertrophy are recorded, and only in 2% they are not accompanied by impaired repolarization [18]. If the voltage is very high, then it is worth suspecting the storage disease [13]. If, on the contrary, the QRS voltage is reduced or normal with severe hypertrophy on ECG, then amyloidosis should be suspected [15] (Figure 4).
Every eighth patient with HCM has elongated QT interval >480 ms, and every second patient has >450 ms, which is associated with the risk of SCD and is an additional argument for implantation of a cardioverter-defibrillator [13][19].
About 5-10% of patients with HCM phenotype have either a normal ECG or an isolated increase in QRS voltage. In such patients, the disease debuts later, the symptoms are less pronounced and the prognosis is better [13][20].
Physiological hypertrophy in athletes does not exceed 14 mm in men [21] and 12 mm in women [22]; nevertheless, it is always suspicious of the HCM onset. One of the most characteristic ECG signs of an athlete’s heart is a pronounced increase in QRS voltage, which is often mistakenly considered as LV hypertrophy. Unlike pathological hypertrophy, there are no concomitant repolarization abnormalities on an athlete’s ECG, therefore, a moderate LV wall thickening on echocardiography in combination with an isolated increase in QRS voltage indicates physiological myocardial remodeling. Repolarization disorders in the form of inverted T wave >1 mm in more than 2 contiguous inferior (II and aVF) and, especially, lateral (I, aVL, V5 or V6) leads indicate a possible HCM [4].
Inverted T wave in the inferior and lateral leads are recorded on the ECG of athletes without structural cardiac changes. Isolated T wave inversions in the inferior leads are found in 2% of white and 6% of healthy black athletes [13], which is much more frequent than inherited heart diseases. According to our unpublished data with 1435 ECGs of athletes from various sports, isolated T wave inversions in the inferior leads are found in 1% of cases. Inverted T waves in the lateral leads are considered the most unfavorable, since they may be the first sign of cardiomyopathy [23]. Abnormalities suspicious of HCM in athletes also include pathological Q waves (>0,25 from the R wave or >40 ms), ST depression >0,5 mm in >2 contiguous leads, complete LBBB, non-specific prolonged QRS >140 ms, and frequent premature ventricular contractions [4].
LV systolic dysfunction
Dilated cardiomyopathy (DCM) is a syndrome characterized by systolic dysfunction and LV dilatation, which cannot be explained by coronary artery disease or conditions leading to LV overload (HTN, valvular and congenital heart disease). LV systolic dysfunction (LVEF <45%) without dilatation since 2016 has been classified as hypokinetic non-dilated cardiomyopathy [24].
DCM is the most etiologically heterogeneous cardiomyopathy. About 40% of DCM cases are inherited [25], which can manifest as isolated heart disease, in combination with conduction defects and noncompacted myocardium (NCM), or within the systemic muscle diseases. Among the latter, the most common DCM phenotype occurs in muscular dystrophies (Duchenne and Becker), limb-girdle muscular dystrophies (LGMD), and Emery-Dreifuss muscular dystrophy (EDMD) [26]. Familial DCM occurs due to mutations in the genes of sarcomere (titin), cytoskeleton (dystrophin, desmin), cell membranes (lamin, ion channels), and organelles [25]. Acquired DCMs develop due to infections, autoimmune diseases, toxic (alcohol, cocaine) or medication (chemotherapy) myocardial damage, micronutrient deficiencies, endocrine and metabolic diseases, and pregnancy [24]. Separately, authors distinguish tachycardia-induced cardiomyopathy — a potentially reversible decrease in LV systolic function, which develops with permanent atrial or ventricular tachyarrhythmia [27]. There is evidence that patients with non-familial DCM also have a genetic substrate of the disease [25][28][29][30].
Table 1
Specific signs of cardiomyopathies on resting ECG
| Phenotype | ECG abnormalities Suggested diagnosis | |
|---|---|---|
| Dilatation/impaired RV contractility | inverted T in V1-V3 | major criterion of right-dominant ACM |
| inverted T in V1-V4(V6) | significant RV involvement | |
| ?-wave in V1-V2 | minor criterion of right-dominant ACM | |
| TAD >55 ms in V1-V3 | minor criterion of right-dominant ACM | |
| ? QRS in limb leads | biventricular ACM | |
| inverted T inV4-V6/I, aVL | biventricular ACM | |
| LV hypertrophy | shortened PQ | Fabry, Danon, Pompe, PRKAG2, mitochondrial diseases |
| AV blocks | amyloidosis, end-stage Fabry, Danon, acute myocarditis | |
| ?? QRS voltage | Danon, Pompe | |
| ? or ’normal’ QRS voltage | amyloidosis | |
| right QRS axis deviation | Noonan syndrome | |
| LV systolic dysfunction | > Р/atrial standstill | type 1 and 2 EDMD |
| sinus bradycardia | laminopathy | |
| shortened PQ | DMD | |
| AV blocks | sarcoidosis, laminopathy, EDMD, myotonic dystrophy, desminopathy, Chagas disease, diphtheria, Lyme disease | |
| Q/QS in inferolateral leads | DMD, BMD, sarcoidosis, LGMD | |
| ? QRS | Left-dominant ACM | |
| Complete RBBB | DMD, Chagas disease (+ left anterior fascicular block) | |
| inverted T in V1-V6 | Left-dominant and biventricular ACM | |
| Hypertrabeculation of LV | Complete LBBB | NCM |
| pathological Q waves | ||
| inverted T | ||
Abbreviations: ACM — arrhythmogenic cardiomyopathy, LV — left ventricle, LBB — left bundle branch, EDMD — Emery-Dreifuss muscular dystrophy, NCM — noncompacted myocardium, RV — right ventricle, LGMD — limb-girdle muscular dystrophies, BMD — Becker muscular dystrophy, DMD — Duchenne muscular dystrophy, TAD — terminal activation delay.
Figure 1. The 42-year-old female patient with biventricular ACM and dominated LV involvement. A. ECG: low QRS voltage in limb leads and inverted T wave in V1-V6; frequent premature ventricular contractions with a configuration of complete LBBB and the superior axis. B. MRI: biventricular dilatation, LV NCM.
Epsilon wave, previously referred to major ACM criteria, is a reproducible low-amplitude signal between the QRS end and T-wave beginning. Over the past ten years, the diagnostic value of this criterion has been questioned due to its various interpretation [7], and in the 2020 updated Padua criteria [5], the epsilon wave is attributed to the minor criterion of right-dominant ACM, as well as terminal activation duration >55 ms, measured from the nadir of the S wave to the end of the QRS, including R’, in V1, V2, or V3 (in the absence of complete RBBB).
Low QRS voltage in limb leads (<5 mm) may indicate LV involvement in ACM (Figure 1). The sensitivity of this criterion is low (<30%); therefore, it is considered minor for the left-dominant ACM in the absence of obesity, emphysema, or pericardial effusion [5]. Also, minor criteria for left-dominant ACM include inverted T waves in left precordial leads (in the absence of complete left bundle branch block (LBBB)) [5]. The isolated left-dominant ACM is phenotypically indistinguishable from dilated cardiomyopathy (DCM) and is often confirmed only by genetic testing.
Registration of late potentials using a signal-averaged ECG has not found wide application in practice and is no longer used for the diagnosis of ACM.
Ventricular arrhythmia with a configuration of complete LBBB and the inferior axis, which indicates its origin from the RV outflow tract is a minor criterion, and without the lower axis, it is a major criterion for right-dominant ACM [5] (Figure 1). Ventricular arrhythmia with a configuration of complete RBBB is a minor criterion for left-dominant ACM [5].
Myocardial changes similar to ACM, such as significant RV enlargement, borderline decrease in the RV ejection fraction (EF), and ventricular arrhythmias, can be induced by regular exercises in healthy individuals [8][9]. The physiological RV changes on the ECG include voltage criteria for RV hypertrophy, isolated complete RBBB, and right axis deviation Highly sensitive to ACM inversed T-wave, in the right-sided chest leads loses its specificity in athletes due to a fairly high prevalence (up to 4% in V1-V3 and 10% in V1-V2) [2]. According to our unpublished data based on the analysis of 619 ECG records of athletes, inversed T-waves in V1-V3 without any significant structural changes in the heart occur in 1,9% of cases (Figure 2). Nevertheless, today in white athletes, any inversed T-waves in two contiguous leads, including in V1-V3, is regarded as pathological and requires in-depth examination and follow-up. In black athletes, such inversions, especially with ST- segment elevation and a J-point >1 mm, are referred to as benign [10]. In addition to inversed T-waves, ACM can be suspected in athletes when recording ventricular arrhythmias and epsilon waves.
Figure 2. A. The 24-year-old male athlete, pentathlon, asymptomatic. Echocardiography, cardiac MRI, 24-hour ECG monitoring did not reveal any pathology. ECG: inverted T-wave in V1-V3 with preceding ST elevation at J-point >1 mm. B. The 35-year-old female athlete, cycling, asymptomatic, without structural heart disease. ECG: inverted T wave in V1-V3, isolated increase in QRS voltage.
LV hypertrophy
Hypertrophic cardiomyopathy (HCM) is the most common inherited heart disease. In 60% of patients, HCM is caused by sarcomere gene mutations. In 5-10%, HCM is simulated by rare storage diseases (Fabry, Danon, PRKAG2 cardiomyopathy), infiltrative diseases (amyloidosis, sarcoidosis), mitochondrial, neuromuscular diseases (Friedreich’s ataxia), malformations (Noonan syndrome), and endocrine cardiomyopathies. In the remaining 30% of patients, the cause of HCM has not yet been clarified [11]. Unlike ACM, the criteria for the HCM diagnosis are only morphological: LV myocardial thickening in adults >15 mm (>13 mm in the presence of a relative with HCM), which cannot be explained by other conditions leading to LV overload (HTN, aortic valve stenosis) [12].
ECG abnormalities, mainly inversed T-waves or deep narrow (“dagger-like”) Q waves with a positive T wave in the inferior and lateral leads, are recorded in more than 90% of patients with sarcomeric HCM [13]. “Giant” (>10 mm) symmetric T waves, usually present in all chest leads, indicate severe hypertrophy of the LV apex [14] (Figure 3). Pseudo-infarct QS complexes in the chest leads and complete bundle branch blocks occur infrequently in HCM and mainly after surgical reduction of the interventricular septum or in severe transmural fibrosis [13]. Such changes are more typical for infiltrative diseases [15][16] (Figure 4).
Figure 3. The 34-year-old male patient with the apical familial HCM. A. ECG: “giant” inverted T-waves in V2-V6, LV hypertrophy voltage signs. B. Echocardiography: hypertrophy of LV apex with ace-of-spades sign.
Figure 4. The 64-year-old female patient with familial transthyretin amyloidosis of the heart. A. ECG: low QRS voltage in limb leads and “normal”, not corresponding to the hypertrophy severity on echocardiography, in the chest leads; R regression in V1-V4. B. Echocardiography: severe LV hypertrophy, atrial dilatation. C. Doppler echocardiography: a restrictive LV diastolic dysfunction.
The combination of visually severe LV hypertrophy with conduction abnormalities on the ECG is always suspicious for HCM phenocopies. Thus, a shortened PQ interval should be suggestive of storage [17] or mitochondrial diseases [1], while an atrioventricular conduction delay suggests amyloidosis [15], sarcoidosis [16], or end-stage storage and mitochondrial diseases [1][13][17].
In many patients with HCM, voltage criteria for LV hypertrophy are recorded, and only in 2% they are not accompanied by impaired repolarization [18]. If the voltage is very high, then it is worth suspecting the storage disease [13]. If, on the contrary, the QRS voltage is reduced or normal with severe hypertrophy on ECG, then amyloidosis should be suspected [15] (Figure 4).
Every eighth patient with HCM has elongated QT interval >480 ms, and every second patient has >450 ms, which is associated with the risk of SCD and is an additional argument for implantation of a cardioverter-defibrillator [13][19].
About 5-10% of patients with HCM phenotype have either a normal ECG or an isolated increase in QRS voltage. In such patients, the disease debuts later, the symptoms are less pronounced and the prognosis is better [13][20].
Physiological hypertrophy in athletes does not exceed 14 mm in men [21] and 12 mm in women [22]; nevertheless, it is always suspicious of the HCM onset. One of the most characteristic ECG signs of an athlete’s heart is a pronounced increase in QRS voltage, which is often mistakenly considered as LV hypertrophy. Unlike pathological hypertrophy, there are no concomitant repolarization abnormalities on an athlete’s ECG, therefore, a moderate LV wall thickening on echocardiography in combination with an isolated increase in QRS voltage indicates physiological myocardial remodeling. Repolarization disorders in the form of inverted T wave >1 mm in more than 2 contiguous inferior (II and aVF) and, especially, lateral (I, aVL, V5 or V6) leads indicate a possible HCM [4].
Inverted T wave in the inferior and lateral leads are recorded on the ECG of athletes without structural cardiac changes. Isolated T wave inversions in the inferior leads are found in 2% of white and 6% of healthy black athletes [13], which is much more frequent than inherited heart diseases. According to our unpublished data with 1435 ECGs of athletes from various sports, isolated T wave inversions in the inferior leads are found in 1% of cases. Inverted T waves in the lateral leads are considered the most unfavorable, since they may be the first sign of cardiomyopathy [23]. Abnormalities suspicious of HCM in athletes also include pathological Q waves (>0,25 from the R wave or >40 ms), ST depression >0,5 mm in >2 contiguous leads, complete LBBB, non-specific prolonged QRS >140 ms, and frequent premature ventricular contractions [4].
LV systolic dysfunction
Dilated cardiomyopathy (DCM) is a syndrome characterized by systolic dysfunction and LV dilatation, which cannot be explained by coronary artery disease or conditions leading to LV overload (HTN, valvular and congenital heart disease). LV systolic dysfunction (LVEF <45%) without dilatation since 2016 has been classified as hypokinetic non-dilated cardiomyopathy [24].
DCM is the most etiologically heterogeneous cardiomyopathy. About 40% of DCM cases are inherited [25], which can manifest as isolated heart disease, in combination with conduction defects and noncompacted myocardium (NCM), or within the systemic muscle diseases. Among the latter, the most common DCM phenotype occurs in muscular dystrophies (Duchenne and Becker), limb-girdle muscular dystrophies (LGMD), and Emery-Dreifuss muscular dystrophy (EDMD) [26]. Familial DCM occurs due to mutations in the genes of sarcomere (titin), cytoskeleton (dystrophin, desmin), cell membranes (lamin, ion channels), and organelles [25]. Acquired DCMs develop due to infections, autoimmune diseases, toxic (alcohol, cocaine) or medication (chemotherapy) myocardial damage, micronutrient deficiencies, endocrine and metabolic diseases, and pregnancy [24]. Separately, authors distinguish tachycardia-induced cardiomyopathy — a potentially reversible decrease in LV systolic function, which develops with permanent atrial or ventricular tachyarrhythmia [27]. There is evidence that patients with non-familial DCM also have a genetic substrate of the disease [25][28][29][30].
Table 1
Specific signs of cardiomyopathies on resting ECG
| Phenotype | ECG abnormalities Suggested diagnosis | |
|---|---|---|
| Dilatation/impaired RV contractility | inverted T in V1-V3 | major criterion of right-dominant ACM |
| inverted T in V1-V4(V6) | significant RV involvement | |
| ?-wave in V1-V2 | minor criterion of right-dominant ACM | |
| TAD >55 ms in V1-V3 | minor criterion of right-dominant ACM | |
| ? QRS in limb leads | biventricular ACM | |
| inverted T inV4-V6/I, aVL | biventricular ACM | |
| LV hypertrophy | shortened PQ | Fabry, Danon, Pompe, PRKAG2, mitochondrial diseases |
| AV blocks | amyloidosis, end-stage Fabry, Danon, acute myocarditis | |
| ?? QRS voltage | Danon, Pompe | |
| ? or ’normal’ QRS voltage | amyloidosis | |
| right QRS axis deviation | Noonan syndrome | |
| LV systolic dysfunction | > Р/atrial standstill | type 1 and 2 EDMD |
| sinus bradycardia | laminopathy | |
| shortened PQ | DMD | |
| AV blocks | sarcoidosis, laminopathy, EDMD, myotonic dystrophy, desminopathy, Chagas disease, diphtheria, Lyme disease | |
| Q/QS in inferolateral leads | DMD, BMD, sarcoidosis, LGMD | |
| ? QRS | Left-dominant ACM | |
| Complete RBBB | DMD, Chagas disease (+ left anterior fascicular block) | |
| inverted T in V1-V6 | Left-dominant and biventricular ACM | |
| Hypertrabeculation of LV | Complete LBBB | NCM |
| pathological Q waves | ||
| inverted T | ||
Abbreviations: ACM — arrhythmogenic cardiomyopathy, LV — left ventricle, LBB — left bundle branch, EDMD — Emery-Dreifuss muscular dystrophy, NCM — noncompacted myocardium, RV — right ventricle, LGMD — limb-girdle muscular dystrophies, BMD — Becker muscular dystrophy, DMD — Duchenne muscular dystrophy, TAD — terminal activation delay.