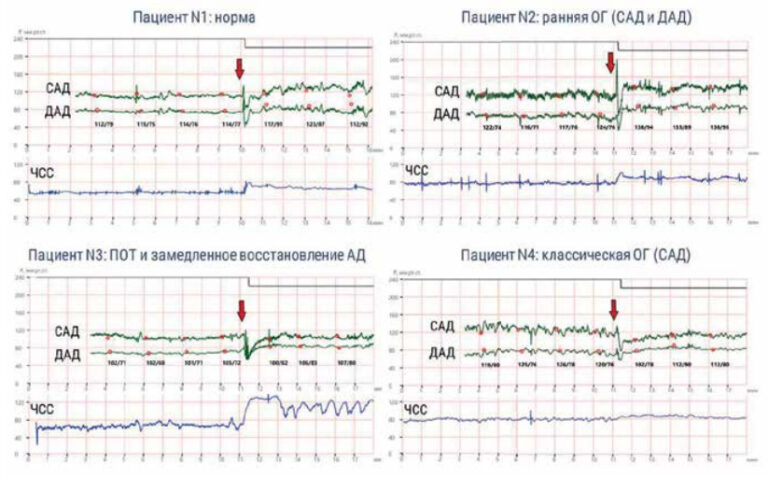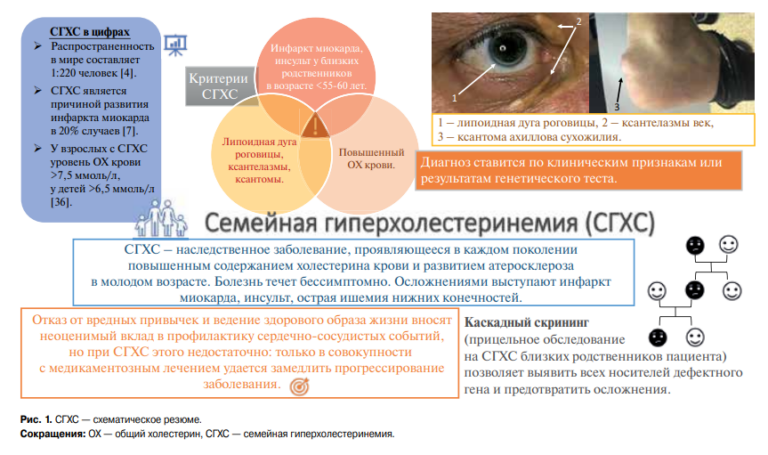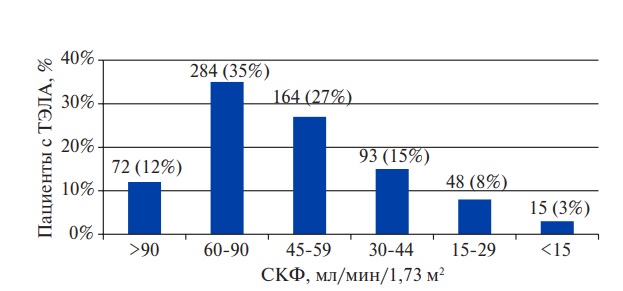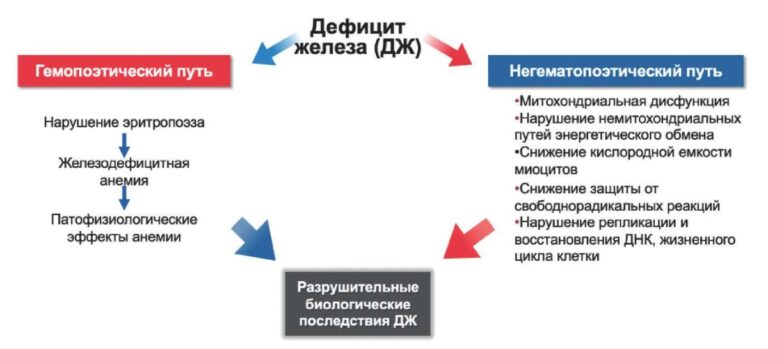
Apical hypertrophic cardiomyopathy as a mask of acute coronary syndrome: a case series
In most cases, the cause of acute coronary syndrome (ACS) is thrombotic coronary artery occlusion. However, under the guise of ACS, patients with various diseases, including with cardiomyopathies, can be admitted to hospital.
Apical non-obstructive hypertrophic cardiomyo pathy (HCM) or Yamaguchi syndrome is a subtype of HCM, the main feature of which is concentric hypertrophy of left ventricular (LV) apex, as a result of which its cavity at the diastole end acquires a peculiar shape resembling an ace of spades. In systole, complete obliteration of the LV apex occurs due to the contact of the contracting parts of the hypertrophied myocardium with each other. This disease was first described by Japanese researchers Sakamoto T, et al. in 1976 and Yamaguchi H, et al. in 1979 [1]. The rare occurrence of this HCM type often erroneously leads to overdiagnosis of ACS and delayed diagnosis. Below are two clinical case reports of patients with apical HCM, which clinically debuted under the guise of ACS.
Forty three-years-old male patients was admitted with suspected non-ST elevation ACS with complaints of pressing retrosternal pain with minimal physical exertion and at rest, lasting up to 30 minutes, radiating to the left arm.
Since 2008, the patient has had an increase in blood pressure (BP), up to a maximum of 200 and 100 mm Hg. In 2015, the patients had myocardial infarction (MI). The patient regularly took only acetylsalicylic acid. Pressing retrosternal pain during physical exertion bothered the patient for a month. Echocardiography, performed 2 days before hospitalization, revealed hypokinesia of the middle anterior, middle anterolateral segments, akinesia of the middle posterolateral, apical septal, apical anterior, apical lateral, apical posterior segments, mo – derate concentric hypertrophy of the LV walls; the Simpson’s ejection fraction (EF) was 45%, and the LV mass index was 125 g/m2. The performed stress echocardiography revealed a decrease in exercise tolerance and a deepening of the initial ST depression in II, III, aVF, V4-V6 up to 1,7 mm. During the last 10 days, angina pain when walking became more frequent, longer, radiating to the left arm, accompanied by sweating, shortness of breath. On the night before the admission, pressing retrosternal pain occurred twice, lasting up to 30 minutes.
At the time of admission, the patients had the state of moderate severity, clear consciousness, the moist and warm skin of normal color. There were muffled heart tones without murmurs. The cardiac rhythm was normal. Heart rate was 65 bpm, BP — 150 and 90 mm Hg. Body mass index was 35 kg/ m2. There were no signs of systemic and pulmonary circulatory congestion. Lung auscultation revealed vesicular breathing without wheezing. The abdomen was soft and nontender. The liver was on the edge of the costal margin. Pasternatsky’s symptom was negative on both sides.
There was negative family history for myocardial infarction, death at a young age, early heart failure in close relatives. An interesting fact is that the patient’s grandfather was Japanese by nationality.
On admission, electrocardiography (ECG) showed ST segment depression in leads I, II, V4-V6, deep ne – gative T waves in leads I, II, AVL, V2-V6, ST elevation in III, signs of LV hypertrophy (Figure 1). Troponin I was 0,40 pg/ml (reference values, 0-0,5 pg/ml). The GRACE score was 90 (low risk). Complete blood count and biochemical tests were within the reference values without unfavorable changes over time.
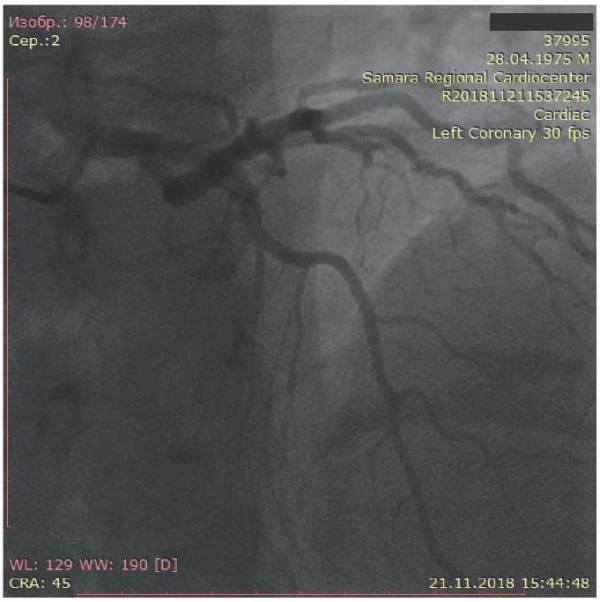
The patient underwent coronary angiography to assess the coronary artery damage and subsequently decide on the need for myocardial revascularization. The left coronary artery had uneven contours. Left anterior descending artery had uneven contours and “muscle bridge” in the middle third of the 2nd segment with stenosis in systole up to 75%. Circumflex artery had uneven contours without hemodynamically significant stenoses (Figure 2). The right coronary artery (Figure 3) had uneven contours and a slight aneurysmal expansion in the middle third of the 1st segment, as well as a 30% stenosis in the middle third of the 3rd segment. The posterior interventricular artery is represented by two branches, one of which departs from the middle third of the 3rd segment, with 30% ostial stenosis.
Echocardiography revealed asymmetric hypertrophy of the LV walls, papillary muscles of the mitral valve with slight intraventricular obstruction (Figure 4) — Yamaguchi variant. The LV weight was 321 g, the mass index was 126 g/m2. LV outlet was 18,8 mm, peak gradient — 7 mm Hg, interventricular septum (IVS) in the apical part in diastole — 15 mm, apex in diastole — 22 mm, anterior wall in the apical segment in diastole — 17 mm, apical-lateral segment in diastole — 14 mm, intraventricular peak gradient at the level of the base of the papillary muscles — 18 mm Hg. Local LV contractility impairment was not detected. The Simpson’s EF was 64%.
Thus, based on the data of investigations, the patient was diagnosed with apical HCM (Yamaguchi syndrome) and was prescribed the following therapy: bisoprolol 5 mg/day, perindopril 5 mg/day.
The second case report. A 46-year-old man complained of an increase in blood pressure to 220 and 120 mm Hg, accompanied by headache, chest discomfort. The condition worsened within two days, when, against the background of high blood pressure, retrosternal discomfort appeared. The patient went to the polyclinic. The ECG revealed ST segment depression in I, II, V4-V6 leads, negative T waves in I, II, AVL, V2-V6 leads. The patient was referred for hospitalization with a diagnosis of non-ST elevation ACS. The patient notes an increase in blood pressure for 20 years with a maximum of 220 and 120 mm Hg. He did not receive antihypertensive therapy regularly. Previously, there were no angina pain in history. The patient was hospitalized in the cardiology department on an emergency basis.
The family history is not burdened, he denies cases of MI, death at a young age, early development of chronic heart failure in close relatives.
At the time of admission, the patients had the state of moderate severity, clear consciousness, the moist and warm skin of normal color. There were muffled heart tones without murmurs. The cardiac rhythm was normal. Heart rate was 81 bpm, BP — 180 and 100 mm Hg. Body mass index was 28,4 kg/ m2. There were no signs of systemic and pulmonary circulatory congestion. Lung auscultation revealed vesicular breathing without wheezing. The abdomen was soft and nontender. The liver was on the edge of the costal margin. Pasternatsky’s symptom was negative on both sides.
There was negative family history for myocardial infarction, death at a young age, early heart failure in close relatives.
ECG at admission revealed ST segment depression up to 1 mm in leads I, II, V4-V6, deep negative T waves in leads I, II, AVL, V2-V6, signs of LV hypertrophy (Figure 5).
High sensitivity troponin I was 0,10 ng/ml (reference values, 0-0,0175 ng/ml). Complete blood count and biochemical tests were within the reference values without unfavorable changes over time.
Echocardiography at admission revealed LV outlet of 18 mm with a peak gradient at rest of 32 mm Hg, with a Valsalva maneuver of 37 mm Hg (Figure 6). In the region of the apex, the wall thickness in diastole was 23 mm, whule the lateral wall in the lower part of the middle third was 23 mm. IVS thickness was 23 mm. There was left atrial dilatation (43 mm). Local LV contractility impairment was not detected. The EF was 68%. There was following conclusion: asymmetric LV hypertrophy of the Yamaguchi type with slight LV obstruction at the level of IVS middle third and papillary muscles of the mitral valve.
Taking into account complaints, anamnestic and echocardiographic data, multislice computed tomography (MSCT) coronary angiography was performed.
According to MSCT coronary angiography, the Agatston score of 2 was revealed. Coronary arteries were not narrowed (CAD-RADS — 0). There was “muscle bridge” of the left anterior descending artery (Figure 7). Based on MSCT data, the diagnosis of non-ST elevation ACS was ruled out, and a diagnosis of apical HCM (Yamaguchi disease) was made. Elevated troponin levels have been associated with hypertensive crisis. In accordance with the diagnosis, the following therapy was prescribed: bisoprolol 5 mg/day, perindopril 10 mg/day, amlodipine 10 mg/day, moxonidine 0,6 mg/day.

Figure 1. Initial ECG of the first patient upon admission to the hospital.

Figure 2. Coronary angiography: left coronary artery of the first patient.

Figure 3. Coronary angiography: right coronary artery of the first patient.

Figure 4. Echocardiography of the first patient. B mode, apical four-chamber view. Hypertrophy of the LV apex, lower IVS segments, anterior, lateral walls, papillary muscles.

Figure 5. ECG of the second patient upon admission to the hospital.

Figure 6. Echocardiography of the second patient. B mode, apical four-chamber view. Hypertrophy of the LV apex, IVS, anterior wall.

Figure 7. 3D reconstruction of the ascending aorta and coronary arteries of the second patient.
Discussion
The prevalence of the apical non-obstructive cardiomyopathy among all patients with HCM is up to 25% in Asia and up to 10% in other countries. Yamaguchi cardiomyopathy is more often diagnosed in men, and the disease onset occurs at the age of 40-60 years [1].
Complaints of patients with this pathology are varied and non-specific, such as cardialgia, which in some patients are angina in nature; shortness of breath during exercise, less often palpitations, dizziness. Cases of severe heart failure and sudden death are extremely rare. In addition, 33-43% have no clinical manifestations of the disease [1, 2].
Physical examination reveals an increase in the apex beat and an abnormal IV or, less commonly, III sound can be detected; the boundaries of relative cardiac dullness, as a rule, are not expanded. Due to the absence of pathognomonic signs, as well as poor clinical symptoms of this disease, the diagnosis is often made due to following ECG abnormalities: inverted T waves of large amplitude (>1 mV) in V4-V6 and I, II, III, aVF, aVL leads, reflecting the increase in the electrical potential of LV apical segments. ST segment depression and signs of LV myocardial hypertrophy are also observed [3]. Due to the rare occurrence and low alertness of doctors regarding the apical HCM, such ECG changes are often erroneously interpreted by clinicians, which leads to overdiagnosis of MI. Such a case report was presented by Russian authors [4]. In this case, transthoracic echocardiography is helpful in making the diagnosis. There are following characteristic ultrasound signs: isolated myocardial hypertrophy of LV apical region; “spike-like” shape of the LV cavity at the end of diastole with a “spike” point in the apex area; the disappearance at the systole end of LV apical part due to the complete contact of the segments of the apex; no signs of subaortic obstruction of the LV outflow tract [3]. The global LV systolic function, assessed by contractility parameters, is supernormal in patients with HCM, which is expressed by a high LVEF [5]. However, in the first clinical observation of the patient, according to echocardiography performed before hospitalization, LV systolic dysfunction was revealed as follows: EF of 45%, hypo- and akinesia areas. Systolic dysfunction could be associated both with LV myocardial ischemia due to severe hypertrophy, and subjective data interpretation by the specialist who performed the investigation.
In the second case report, according to echocardiographic data, the patient has both asymmetric hypertrophy of the IVS and LV apex area. This type of HCM is classified as a mixed type of apical HCM [3]. The patient also has an obstruction at the level of the middle third of the IVS with a peak gradient at rest of 32 mm Hg, with a Valsalva maneuver of 37 mm Hg. According to the current clinical guidelines of the Russian Society of Cardiology for HCM, pressure gradient criteria for midventricular obstruction have not been developed [5]. Thus, we regarded this case as an apical type of HCM.
When visualizing the coronary arteries in patients, a “muscle bridge” was detected in the left anterior descending artery. In most cases, with this anomaly, there are no clinical symptoms, and the pathology remains undiagnosed. However, under conditions of myocardial hypertrophy, there is a relative insufficiency of coronary blood flow, and the presence of a muscle bridge can exacerbate the discrepancy between myocardial perfusion and its needs.
In most cases, ECG and transthoracic echocardiography make it possible to make a correct diagnosis. However, sometimes recognition of apical HCM by echocardiography is difficult, because the LV apex is less accessible for visualization due to near field artifacts. In case of doubt, intravenous ultrasound contrast agents should be used for endocardial contouring. Verification of the apical HCM can also be done using ventriculography or cardiac magnetic resonance imaging. At the same time, a characteristic LV narrowing in the lower third with a sharpening in the apex is revealed, which confirms the data obtained by echocardiography [3].
Currently, there are no clear guidelines for the diagnosis, family screening and risk stratification of patients with apical HCM. According to the 2020 AHA/ACC guidelines for the diagnosis and treatment of patients with HCM, beta-blockers, verapamil, diltiazem are recommended for the treatment of angina syndrome in patients with non-obstructive HCM in the absence of obstructive coronary artery disease [6].
Yamaguchi cardiomyopathy has a less favorable prognosis than previously thought. In a study by Klarich KW, et al. 193 patients with apical HCM were included and compared with age-matched and sex-matched patients without HCM [1]. The 20-year survival rate in patients with apical HCM was significantly worse — 47% vs 60%. Unfavorable prognostic factors were age, female sex and initial atrial fibrillation.
Study limitations. In these cases, contrast-enhanced cardiac magnetic resonance imaging was not performed, because the diagnosis was established by transthoracic echocardiography with the exclusion of coronary artery pathology.
Conclusion
The presented case reports demonstrate a variant of the course of apical HCM with the leading clinical syndrome of chest pain, which can often be the cause of ACS overdiagnosis.
Relationships and Activities: none.

Чтобы читать статью войдите с логином и паролем от scardio.ru
Keywords
For citation
Goncharova D.Yu., Bikbaeva G.R., Tukhbatova A.A., Mullova I.S., Duplyakov D.V. Apical hypertrophic cardiomyopathy as a mask of acute coronary syndrome: a case series. Russian Journal of Cardiology. 2022;27(4S):5262. https://doi.org/10.15829/1560-4071-2022-5262
Copy