Роль внеклеточного матрикса сердца в возникновении и прогрессировании хронической сердечной недостаточности
Сердечно-сосудистая патология лидирует в структуре заболеваемости и смертности всех стран мира. Такие позиции объясняются, в т.ч., появлением и прогрессированием хронической сердечной недостаточности (ХСН) — сложного клинического синдрома, реализующегося вследствие формирования функционального и морфологического ремоделирования миокарда [1]. Интенсивность ремоделирования определяется рядом факторов, важным из которых является реактивность соединительнотканного компонента сердца — внеклеточного матрикса (ВКМ) [2].
ВКМ сердца составляют структурные и неструктурные протеины, которые в зависимости от своего строения и структурной локализации могут выполнять разные функции. Основными структурными белками являются гликопротеины: эластин, фибронектин, ламинин и коллагены I и III типов [3]. Аминокислотная последовательность полипептидных цепей этих белков позволяет сформировать структуру с уникальными механическими свойствами, которая обладает огромной прочностью и эластичностью [4]. Неструктурные белки (протеогликаны) обеспечивают функционирование ВКМ как информационного хаба, накапливающего и транспортирующего сигнальные данные для всех клеток органа. Помимо прочего, протеогликаны создают своеобразный резервуар для факторов клеточного роста, обеспечивающий адаптивно-регенераторные и пластические возможности сердца [5].
Принято считать, что активную роль в реорганизации ВКМ и формировании миокардиального фиброза играют миокардиальные фибробласты. Число этих клеток резко возрастает вследствие эпителиально-мезенхимального перехода, инициирующегося выделяемыми при повреждении агентами — эндотелином-1, трансформирующим фактором роста-? (TGF-?), ангиотензином II [6]. Доказано, что фибробласты сердца являются механосенситивными клетками, имеющими ионные каналы, реагирующие на сжатие и растяжение. Перегрузка камер давлением вызывает в этих клетках раннюю активацию матрикс-синтетической программы в фибробластах, которая реализуется активным синтезом структурных и внеклеточных протеинов ВКМ [7]. Большое значение для инициации фиброза отводится активации нейрогуморальных факторов и окислительному стрессу, вызывающим патологическое ремоделирование миокарда [8].
Установлено, что ВКМ имеет систему саморегуляции, на которую оказывает влияние ряд гемодинамических [9], нейрогуморальных и метаболических факторов [10], реализующихся при ХСН. Изменения биохимического профиля ВКМ могут выступать в качестве патогенетических механизмов ХСН как с сохраненной [11], так и со сниженной фракцией выброса левого желудочка (ЛЖ) [12]. Морфологическая перестройка миокарда у пациентов с ХСН часто становится причиной дебюта предсердных и желудочковых аритмий, которые, в свою очередь, приводят к декомпенсации ХСН [13]. При этом большинство заболеваний, ассоциированных с этим состоянием (артериальная гипертензия, сахарный диабет, ишемическая болезнь сердца, хроническая болезнь почек), способны инициировать триггеры миокардиального фиброза (перегрузка камер сердца, ишемия миокарда, метаболическое повреждение и пр.), что формирует порочный круг.
Одной из ключевых проблем в лечении больных ХСН является необходимость частых госпитализаций или обращений за медицинской помощью в связи с прогрессированием сердечной недостаточности (СН) [14]. Очевидно, что течение ХСН носит индивидуальный характер, что затрудняет стратификацию кардиоваскулярного риска. Оценка состояния ВКМ сердца может стать важным инструментом оценки течения ХСН, предоставляющим дополнительную информацию для выбора тактики ведения конкретного больного (необходимость стационарного лечения, коррекция медикаментозной терапии, имплантация кардиовертера-дефибриллятора, проведение ресинхронизирующей терапии, использование устройств механической поддержки ЛЖ, трансплантация сердца).
Роль ВКМ в развитии и прогрессировании СН
Формирующаяся вследствие острой ишемии и/или активного воспаления травма кардиомиоцитов инициирует структурное и функциональное ремоделирование миокарда с исходом в гипертрофию и/или замещение экстрацелюлярного матрикса соединительной тканью [15]. В основе такой морфологической перестройки миокарда лежит возникшее нарушение устойчивого баланса между синтезом, метаболизмом и деградацией белков ВКМ сердца. Прогрессирующее нарастание жесткости миокарда обоих желудочков приводит сначала к диастолической, а впоследствии и к систолической дисфункции сердца.
В настоящее время не вызывает сомнений наличие связи между прогрессией миокардиального фиброза и возникновением ХСН. Более того, установлено, что ВКМ сердца играет важную роль в патофизиологии декомпенсации ХСН [16]. Изначально возникающее ремоделирование миокарда является компенсаторным механизмом, направленным на обеспечение адекватного сердечного выброса и нормализацию систолического и диастолического напряжения стенок ЛЖ [17]. В случае невозможности обеспечить нормальную тканевую перфузию при изменившихся условиях (нагрузка объемом и/или давлением, ишемия) ранее инициированное ремоделирование ВКМ становится причиной сначала клеточной, а потом и органной дисфункции.
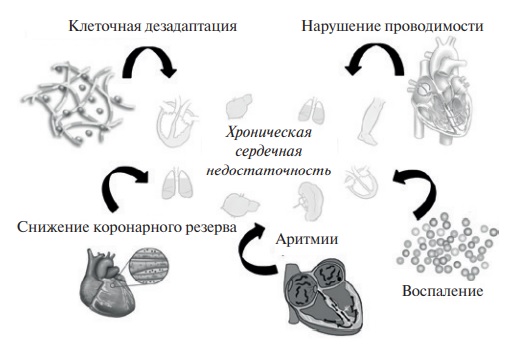
В большинстве случаев рассматриваемые ниже патофизиологические сценарии могут выступать как в качестве факторов, инициирующих ХСН, так и приводить к декомпенсации сердечной деятельности (рис. 1).

Рис. 1. Механизмы возникновения и прогрессирования ХСН, ассоциированные с состоянием ВКМ.
Электромеханическая диссинхрония
Известно, что систолическая функция сердца обеспечивается согласованным сокращением камер, которое происходит вследствие распространения электрических импульсов по элементам проводящей системы сердца. В случае нарушения внутрипредсердной, предсердно-желудочковой, внутрижелудочковой проводимости формируется соответствующий паттерн диссинхронии, ассоциированный с СН [18]. Высказывается мнение о том, что изменённая геометрия ВКМ сердца способствует хаотичному разобщению ранних и поздно-активируемых участков миокарда, что может вызывать или усугублять уже имеющиеся нарушения проводимости, ослабляя сократимость и замедляя скорость проведения электрических импульсов [19].
Чаще всего у больных ХСН миокардиальный фиброз приводит к блокаде левой ножки пучка Гиса, характеризующейся аномальной активацией желудочков. Вследствие того, что электрический импульс распространяется эксцентрично и не по системе Гиса-Пуркинье, а непосредственно по миокарду, он достигает ЛЖ позднее. Фазы быстрого и медленного наполнения желудочков наслаиваются друг на друга, уменьшая вклад предсердной систолы. Разобщенная активация папиллярных мышц митрального клапана ведет к поздней диастолической или пресистолической регургитации. Такое электромеханическое ремоделирование отрицательно влияет на системную гемодинамику и способствует развитию СН.
Нарушение кровоснабжения миокарда
Дисбаланс между синтезом и распадом элементов ВКМ может служить причиной возникновения ХСН, даже при сохраненной либо малоизмененной функции кардиомиоцитов, что связывают с их сдавлением избыточно разросшейся коллагеновой сетью. Такая морфологическая перестройка увеличивает жесткость миокарда при растяжении, приводит к диастолической дисфункции и ухудшению кровоснабжения миокарда [20].
Помимо этого, развивающийся реактивный фиброз может приводить к аккумуляции фиброзной ткани в периваскулярном пространстве вокруг интрамуральных коронарных артерий [21]. Описаны клинический ситуации, когда повреждение ВКМ приводило к дестабилизации и разрыву атеросклеротических бляшек [22]. У больных с неишемической кардиомиопатией периваскулярный коронарный фиброз значительно снижает коронарный резерв, что также может становиться причиной декомпенсации ХСН [23].
Нарушение межклеточных контактов
В норме пространственное расположение клеток определяется белками ВКМ. При нарушении его структуры кардиомиоциты утрачивают связь со своим окружением и покидают свое изначальное место. Ответом организма на такую “миграцию” является активация механизма аноикиса, по сути представляющего собой разновидность апоптоза. Как и классический апоптоз, аноикис может запускаться внутренним образом, через повреждение митохондрий, и внешним, в ответ на активацию поверхностных “рецепторов смерти” [24]. Olivetti G, et al., исследовав 36 сердец после трансплантации этого органа, обнаружили, что интенсивность апоптоза в миокарде больных ХСН была в 232 раза выше по сравнению с контрольной группой пациентов, прижизненно не имевших СН [25]. Предполагается, что в роли активаторов аноиксиса могут выступать некоторые профиброгенные биологические агенты и провоспалительные цитокины, а одним их возможных механизмов реализации может выступать оксидативный стресс [26]. Таким образом, наряду с нейрогуморальным и гемодинамическим эффектами миокардиального фиброза, в формировании ХСН могут принимать участие и механизмы клеточной дезадаптации [27].
Активация медиаторов воспаления
В 1990г после выявления в крови больных ХСН высокого уровня циркулирующих провоспалительных цитокинов впервые заговорили о наличии тесной патофизиологической связи между СН и воспалением [28]. Последующие исследования только укрепили это убеждение и позволили сформулировать “цитокиновую” модель патогенеза ХСН [29]. Среди возможных цитокин-опосредованных механизмов развития ХСН исследователи отмечают снижение экспрессии генов, регулирующих кальциевый обмен [30], инициацию гипертрофии и повышение “жесткости” миокарда [31], активацию апоптоза и формирование зон с нарушенной электрической проводимости [32].
Цитокины, индуцирующие ремоделирование сердца и предопределяющие формирование СН, могут синтезироваться кардиомиоцитами (кардиокины) и сердечными фибробластами [33] в ответ на перерастяжение. В связи с этим высказываются предположения, что не только воспаление может приводить к СН, но и сама ХСН имеет потенциал для реализации цитокин-опосредованного повреждения миокарда [34]. По всей видимости, при таком развитии событий ВКМ выполняет роль “мессенджера” между инициирующими факторами и миокардом, результатом чего является возникновение либо декомпенсация ХСН.
Аритмогенный сценарий ХСН
Одним из факторов, ухудшающих течение ХСН, является повышение частоты предсердных нарушений ритма сердца, в частности фибрилляции предсердий (ФП). Было установлено, что не только перестройка ВКМ приводит к возникновению и хронизации ФП, но и сама аритмия имеет профибротический потенциал [35]. Основой реализации этого потенциала является анизотропное проведение возбуждения, возникающее вследствие сепарации интактных мышечных волокон соединительной тканью [36]. Сердечные фибробласты, изначально электрически невозбудимые клетки, могут выступать в качестве информационных мостиков между кардиомиоцитами, поддерживая электрическое ремоделирование миокарда при ФП. Результатом таких взаимодействий является изменение локальных рефрактерных периодов миокарда предсердий и индукция спонтанной диастолической деполяризации [37].
Патофизиологические основы причинно-следственной связи между ХСН и ФП до конца не определены: в большинстве случаев не удается сделать однозначный вывод о том, являлась ли ФП причиной декомпенсации ХСН, либо была ее следствием. Между тем, не вызывает сомнений, что сочетание ХСН и ФП значительно увеличивает частоту госпитализаций по причине декомпенсации сердечной деятельности [38].
Диагностика нарушения ВКМ при СН
Исследование процессов структурно-функциональной реорганизации ВКМ сердца может позволить составить целостное представление о механизмах ремоделирования миокарда при ХСН. Между тем, унифицированного диагностического алгоритма для оценки состояния ВКМ в настоящее время не разработано, а имеющиеся методики не всегда находят применение в повседневной клинической практике.
Эндомиокардиальная биопсия является золотым стандартом диагностики фиброза миокарда, но имеет ряд ограничений и рисков, свойственных инвазивным методам диагностики, что ограничивает широкое использование в клинике. Кроме того, при получении биоптата исследуют локальный участок миокарда, который не всегда отражает морфологическое состояние исследуемой камеры и тем более всего сердца [39]. Более доступными и легко воспроизводимыми являются методики неинвазивной диагностики фиброза миокарда.
Одной из самых высокочувствительных методик, позволяющих детектировать патологические скопления коллагена, является магнитно-резонансная томография сердца [40]. Основу заключения составляет исследование зон отсроченного накопления хелатных солей гадолиния — late gadolinium enhancement (LGE). Будучи инертным внеклеточным агентом, это вещество способно проникать через мембрану кардиомиоцитов только при их повреждении, поэтому при постинфарктном кардиосклерозе и фиброзе любой этиологии возникает повышенное накопление этого контрастного препарата [41]. Перспективным инструментом оценки состояния ВКМ сердца считается Т1 картирование. Суть метода заключается в определении Т1-времени продольной релаксации ткани и построении на основе полученных значений цветных карт миокарда. При совмещении пиксельных карт до и после контрастирования можно рассчитать внеклеточный объём (ECV), отражающий степень интерстициального фиброза [42].
Ценную информацию о состоянии ВКМ могут предоставить современные ультразвуковые методы исследования сердца. Так, тканевая допплерография позволяет оценить степень изменения толщины или длины участка миокарда от конечной диастолической до конечной систолической величины в процентах — так называемую деформацию или стрейн. Стрейн эластография позволяет измерить эластичность миокарда, т.е. косвенно оценить коллагеновый и эластиновый компоненты ВКМ [43]. Speckletracking эхокардиография отслеживает траекторию движения (tracking) акустических маркеров миокарда (speckle) в ходе сердечного цикла, после компьютерной обработки траектории движения акустических пятен получают цифровые значения, графики и диаграммы деформации, скорости деформации ЛЖ (глобальная деформация) и его сегментов (региональная деформация) в продольном, циркулярном, радиальном направлениях [44]. В ряде исследований было показано, что уменьшение глобальной продольной деформации коррелирует с увеличением риска возникновения неблагоприятных сердечно-сосудистых событий у больных ХСН, позволяет оценивать вероятность развития декомпенсации сердечной деятельности [45].
Расшифровка механизмов перестройки ВКМ путем изучения молекулярных индукторов, отвечающих за эти процессы, является перспективным направлением изучения пространственной кардиоваскулярной цитоархитектоники. Так, было продемонстрировано, что у пациентов ХСН концентрации профиброгенных биологических агентов, циркулирующих в крови, могут отражать риск негативных клинических событий, связанных с развитием фиброза [46]. В связи с этим на протяжении последних нескольких десятилетий повышается интерес к матриксным металлопротеиназам (matrix metalloproteinases — MMP) — цинкзависимым эндопептидазам, участвующим в деградации базальной мембраны и ВКМ. Они секретируются фибробластами, кардиомиоцитами и лейкоцитами в виде неактивных проферментов, а в межклеточном пространстве активируются под действием других протеаз. MMP участвуют в процессах ремоделирования ВКМ, денатурируя фибриллы коллагена [1], а также разрушая такие его компоненты, как эластин, фибронектин, гликозаминогликаны.
Протеолитическая активность MMP в физиологических условиях регулируется специфическими тканевыми ингибиторами матриксных металлопротеиназ (tissue inhibitor of metalloproteinases — TIMP). TIMP продуцируются и секретируются фибробластами, эпителиальными клетками и эндотелиальными клетками и распределяются среди тканей. В настоящее время отмечена связь 4 типов TIMP с фиброзом миокарда, при этом чаще всего такая ассоциация обнаруживается применимо к TIMP-1. Этот фермент опосредует взаимодействие между белком мембраны фибробластов CD63 и интегрином ?1, который широко экспрессируется в различных клетках, вызывая фиброз [8]. Было продемонстрировано, что TIMP-1 может использоваться в качестве сильного предиктора общей двухлетней летальности: высокая концентрация этого биомаркера у больных ХСН в 8 раз увеличивает риск смертельного исхода [47].
Проведенные исследования показали, что при заболеваниях сердечно-сосудистой системы в сыворотке крови нарушается нормальное соотношение MMP и TIMP, повышается количество провоспалительных цитокинов (фактор некроза опухоли-?, интерлейкин-1, С-реактивный белок, галектин-3), запускающих синтез MMP [48]. Это приводит к прогрессированию фиброза и формированию систолической и диастолической дисфункции.
В контексте изучаемого вопроса определенный интерес представляет исследование содержания в крови галектина-3. Этот биомаркер играет важную роль в регуляции процессов воспаления, иммунного ответа, дегенерации нервной ткани и фиброза. Галектин-3 секретируется во внеклеточное пространство в месте повреждения, активирует фибробласты и способствует развитию фиброза. Была выявлена зависимость между уровнем галектина-3 и течением ХСН: повышение уровня галектина-3 указывает на прогрессирование ХСН, а при снижении его концентрации — на уменьшение тяжести заболевания [49]. Эти данные позволяют предположить, что галектин-3 может иметь прогностическое значение в отношении развития нежелательных событий у пациентов, страдающих ХСН.
В крупном исследовании PARADIGM-HF (Prospective Comparison of ARNI With ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure trial) помимо прочего исследовался широкий спектр биомаркеров фиброза: плазменные концентрации альдостерона, TIMP-1, MMP-2 и MMP-9; а также уровни sST2, Gal-3, PINP и PIIINP в сыворотке крови. Все исследуемые показатели находились вне референсных значений, указывая на наличие фиброза миокарда у исследуемых больных с ХСН. Авторы обнаружили значимую связь между исходными уровнями sST-2, TIMP-1, PIIINP и конечными композитными точками исследования (сердечно-сосудистая летальность и госпитализация вследствие декомпенсации ХСН) [50].
Несмотря на представленные данные, в настоящее время, к сожалению, не существует “идеального” биомаркера фиброза, который бы одновременно отражал наличие и степень ремоделирования ВКМ и представлял ценность для стратификации риска неблагоприятного течения ХСН.
Заключение
Имеющиеся данные позволяют утверждать, что нарушение структуры ВКМ сердца является важным патофизиологическим механизмом развития и прогрессирования СН. Понимание такой роли ВКМ и разработка алгоритмов верификации индивидуального статуса ВКМ у пациентов с сердечно-сосудистой патологией способны предоставить дополнительную информацию о течении ХСН, помогут оценить риск развития неблагоприятных сердечно-сосудистых событий и эффективно контролировать проводимую фармакологическую и немедикаментозную терапию.

Чтобы читать статью войдите с логином и паролем от scardio.ru
Ключевые слова
Для цитирования
Илов Н.Н., Арнаудова К.Ш., Нечепуренко А.А., Ясенявская А.Л., Башкина О.А., Самотруева М.А. Роль внеклеточного матрикса сердца в возникновении и прогрессировании хронической сердечной недостаточности. Российский кардиологический журнал. 2021;26(2S):4362. https://doi.org/10.15829/1560-4071-2021-4362
Скопировать