Серия редких клинических случаев наблюдения пациентов с семейной гиперхолестеринемией
Введение в проблему
Семейная гиперхолестеринемия (СГХС) — наиболее частое аутосомно-доминантно наследуемое заболевание, обусловленное мутацией в генах рецептора липопротеинов низкой плотности (LDLR), аполипопротеина В-100 (ApoB) и пропротеинконвертазы субтилизин/кексин 9-го типа (PCSK9) [1][2], приводящее к повышению уровня липопротеинов низкой плотности (ЛНП) в крови, раннему атеросклеротическому поражению сосудов и сердечно-сосудистым событиям в молодом возрасте (рис. 1) [3]. Гомозиготная форма СГХС относится хотя и к тяжелым, но редким наследственным заболеваниям, в то время как гетерозиготная СГХС встречается достаточно часто. По последним оценкам, распространенность СГХС в мире составляет 1 на 220 человек [4]. В Западной Сибири — еще выше — 1:108 [5]. По-прежнему <1% населения в разных странах мира имеет верифицированный диагноз и получает лечение [3]. Хотя раннее начало терапии позволяет снизить степень прогрессирования атеросклероза (АС), в большинстве случаев заболевание диагностируется уже после развития осложнений: СГХС приводит к инфаркту миокарда (ИМ) в возрасте <45 лет в 20% случаев [6][7]. На момент диагностики СГХС до 30% пациентов уже имеют в анамнезе вмешательство на коронарных артериях [8].

Рис. 1. СГХС — схематическое резюме.
Сокращения: ОХ — общий холестерин, СГХС — семейная гиперхолестеринемия.
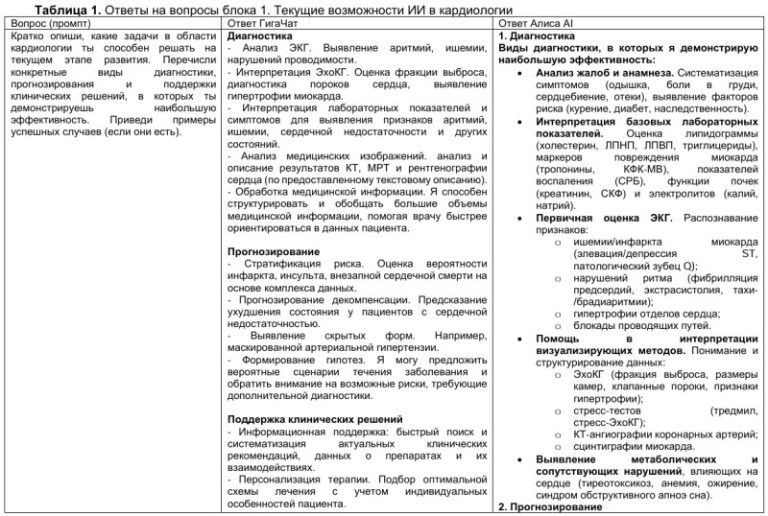
К сожалению, даже те пациенты, кто знает о своей патологии, часто не достигают безопасного уровня холестерина. По данным крупного европейского метаанализа, включившего работы 2006-2017гг (отобранных работ — 81, 303534 пациента высокого и очень высокого риска), где также исследовались пациенты с СГХС (11 работ, 41594 пациента), лишь 35% получали терапию статинами, и только 15% (9-22%) достигали ЛНП <2,5 ммоль/л [9]. По данным российского регистра “РЕНЕССАНС”, включившего 1208 больных СГХС со средним уровнем ЛНП 6,6±2,1 ммоль/л, удельный вес пациентов, принимающих гиполипидемическую терапию, составил 72%, в высокоинтенсивном режиме — 61%. Уровня ЛНП <2,5 ммоль/л достигли 13%, а уровня <1,8 ммоль/л — всего 6% [10]. Отсутствие достижения целевых показателей связано как с недостаточной приверженностью к терапии, так и с малой долей пациентов, получающих высокоинтенсивную терапию с ингибитором PCSK9. По данным регистра “PLANET” Чехии и Словакии (n=1755), среди больных СГХС, принимающих ингибиторы PCSK9 (n=55), 61,8% достигли целевого уровня ЛНП [11]. По данным того же регистра, сердечно-сосудистые события возникали лишь в 14% случаев у пациентов с целевым значением ЛНП.
Клинические случаи СГХС
В настоящее время методика генетического исследования с целью выявления конкретных мутаций, ответственных за СГХС, стала все более доступна на территории России. Несмотря на дороговизну исследования, данные, полученные в результате секвенирования генома, дают полезную дополнительную информацию, глубже раскрывая суть проблемы конкретного индивида и позволяя определить степень “агрессии” в достижении целевых значений ЛНП. Кроме того, знания о типе мутации зачастую позволяют сделать выбор в пользу той или иной гиполипидемической терапии.
Случай 1. Нами наблюдаются мать (1961г рождения) и сын (1990г) с подтвержденным диагнозом СГХС. У обоих отсутствуют сердечно-сосудистые события в анамнезе. У матери выявлено повышение общего холестерина (ОХ) до 7,3 ммоль/л (ЛНП 6,2 ммоль/л). У сына в течение жизни максимальный уровень ОХ составил 7,86 ммоль/л (ЛНП 5,65 ммоль/л). Оба пациента принимают статины, на фоне чего удалось добиться значимого снижения значений ЛНП: у матери ~2,3 ммоль/л на фоне терапии розувастатином, у сына 2,49 ммоль/л (розувастатин 20 мг). При проведении генетического исследования выявлено наличие одинаковой гетерозиготной мутации гена AроB (NP_000375.2:p. Arg3527Gln/NC_000002.11:g.21229160C>T), локализуемой в 26 экзоне. Данный вариант является наиболее частым дефектом, обнаруживаемом в гене AроB, с общей распространенностью 6-10% среди лиц с СГХС [12]. Различные исследования показали, что носители этого варианта, у которых в результате развилась гиперхолестеринемия, имели более мягкий фенотип с точки зрения уровня липидов и возраста развития ишемической болезни сердца (ИБС), чем носители мутации LDLR дефектного типа [13]. Средний возраст начала ИБС (в отсутствии лечения) составляет 50 лет. Достижение показателей ЛНП у сына <2,5 ммоль/л (причем с 22-летнего возраста), т.е. показателя нормального для человека без СГХС, позволяет надеяться, что возраст наступления возможного сердечнососудистого события будет отложен как минимум до 60-65-летнего возраста.
Примечателен тот факт, что, помимо патологической мутации AроB, у матери дополнительно была обнаружена мутация гена PCSK9 (NP_777596.2:p. Arg46Leu/NC_000001.10:g.55505647G>T), обладающая протекторным эффектом и отсутствующая у сына. Данная мутация представляет собой широко изученный полиморфизм [14], который действует как генетический модификатор уровня ЛНП и ответа на статины. Наличие полиморфизма p.Arg46Leu в PCSK9 ассоциировано со значительным снижением уровней ЛНП по сравнению с контрольной группой и меньшей распространенностью сухожильных ксантом [15]. Самое важное, что распространенность ?1 сердечнососудистого события у носителей варианта p.Arg46Leu составляла 11% по сравнению с 33% в контроле, и это различие было статистически значимым (р=0,05). Таким образом, генетическое исследование данной семьи указывает на необходимость раннего начала терапии статинами у носителей мутации AроB и выявляет потенциально лучший отдаленный прогноз у матери, являющейся носителем протекторного гена PCSK9.
Случай 2. Мужчина 41 года с анамнезом проведения чрескожных коронарных вмешательств (ЧКВ) в возрасте 27 и 33 лет с имплантацией суммарно девяти стентов. Максимальный уровень ОХ при выявлении в 27-летнем возрасте составлял 14 ммоль/л. В семейном анамнезе обращает внимание ранняя смерть отца от повторного ИМ в возрасте 57 (первый инфаркт в 27 лет). При выполнении генетического исследования выявлена гомозиготная мутация AроB (NP_000375.2:p.Arg3527Gln/NM_000384.2:c.10580G >A/NC_000002.11:g.21229160C>T). На фоне терапии аторвастатином в дозе 80 мг удалось снизить уровень ОХ до 7,88 и ЛНП до 6,02 ммоль/л, в дальнейшем добавлена комбинация с эзетимибом в дозе 10 мг и эволокумабом 140 мг. Как видно в сравнении с примером 1, гомозиготный тип наследования обуславливает тяжесть течения атеросклероза, исходно более высокие уровни ОХ и ЛНП и необходимость комбинированной холестерин-снижающей терапии для достижения целевых значений ЛНП.
Случай 3. Молодая женщина, 2000г рождения с отягощенным семейным анамнезом (у деда по отцовской линии внезапная сердечная смерть в 44 года, у отца подтвержденный АС коронарных артерий без показаний к реваскуляризации в 41 год) и повышением ОХ до 10,26 ммоль/л и ЛНП до 8,5 ммоль/л. Проведенный генетический анализ показал наличие мутации гена LDLR (NM_000527.4:c.2389+5G>C/ NC_000019.9:g.11238766G>C ) I типа (“нулевая” мутация). Данная мутация ранее встречалась лишь в 2-х случаях у пациентов из России в библиотеке лаборатории “Health in code” (Ла Корунья, Испания). Тем не менее наличие мутации гена LDLR обуславливает крайне высокий риск развития раннего атеросклероза и, возможно, неоптимальный эффект от приема стандартной холестерин-снижающей терапии. Так, у отца пациентки (вероятнее всего имеющего такую же мутацию) на фоне высокоинтенсивной терапии статинами (розувастатин 40 мг) удалось снизить уровень ОХ до 7,24 ммоль/л и ЛНП до 4,95 ммоль/л (исходные документированные значения общего холестерина ~12 ммоль/л). Сама пациентка в настоящее время воздерживается от приема холестерин-снижающих препаратов.
Случай 4. Нами наблюдается семья, 6 членов которой имеют “определенную” СГХС в соответствии с критериями Dutch Lipid Clinic Network (DLCN) (рис. 2). Первым в поле зрения попал молодой мужчина 34 лет (пробанд), у которого ИБС манифестировала ИМ, по поводу которого ему было проведено ЧКВ. Липидограмма выявила повышение ОХ до 8,65 ммоль/л и ЛНП до 6,6 ммоль/л. При подробном сборе анамнеза выяснилось, что родной брат пациента умер от инфаркта в возрасте 32 лет; мать имеет высокий класс стенокардии в возрасте 55 лет; родной дядя по материнской линии перенес ЧКВ в возрасте 43 лет; дед по материнской линии умер в 42 года от ИМ. Все указанные члены семьи имели документированный уровень ОХ >7,5 ммоль/л. Кроме того, дочь пробанда имеет уровень ОХ 9 ммоль/л. Матери пробанда через 1 мес. после выявления СХГС было выполнено аортокоронарное шунтирование по поводу критического поражения трех коронарных бассейнов. Данный пример демонстрирует важность каскадного скрининга и возможность применения клинических критериев для постановки диагноза СГХС и выявления группы риска по раннему развитию сердечно-сосудистых событий.

Рис. 2. Родословная пациента с тяжелой гиперхолестеринемией.
При подготовке материалов данной статьи пациент из примера № 2 умер от повторного сердечнососудистого события. Наблюдение за остальными пациентами продолжается.
Обсуждение
Скрининг
Во всем мире имеется организованная система ранней диагностики наследственных заболеваний, таких как фенилкетонурия, врожденный гипотиреоз и др., целью которой является предотвращение отдаленных и необратимых последствий нарушений обмена. Атеросклеротическое поражение сосудов, развивающееся вследствие неправильного метаболизма липидов, прогрессирует незаметно годами, а осложнения в виде инфарктов и инсультов ложатся тяжелым бременем не только на отдельно взятую семью, но и государство в целом, особенно когда речь идет о трудоспособном населении. Совокупный экономический ущерб от гиперхолестеринемии в Российской Федерации определен в 1295 трлн руб. в год, включая прямые затраты (оказание неотложной помощи, выплаты пособий по инвалидности) и непрямые потери в экономике (потери заработка из-за временной нетрудоспособности; потери внутреннего валового продукта в связи с инвалидностью; потери внутреннего валового продукта, связанные с преждевременной смертностью в экономически активном возрасте) [16]. Продолжительность жизни мужчин с гетерозиготной СГХС в нашей стране не так давно составляла 53 года, женщин — 62 года [17].
С каждым годом в России больше пациентов с тяжелыми формами нарушения липидного обмена включается в регистры, открываются новые липидные центры, однако пока рано говорить об отработанной системе выявления и наблюдения пациентов с СГХС. Этому существует множество причин: недостаточная осведомленность врачей, низкая приверженность к терапии пациентов, отсутствие липидных центров и низкая доступность генетических методов диагностики в регионах. Определенные трудности возникают с выбором скринингового метода для выявления пациентов с СГХС. Хотя гиперхолестеринемия является самостоятельным фактором риска развития АС, изолированное повышение холестерина не позволяет с высокой долей вероятности предсказывать наличие СГХС. К тому же недавние работы не продемонстрировали высокой частоты выявления СГХС в популяции пациентов с тяжелой гиперхолестеринемией (0,3-1,7%) [18][19], а значит экономической эффективности такого скрининга. Wald DS, et al. (2007) предложили измерять уровень ОХ всем детям 1-9 лет, т.к. оказалось, что именно в этом возрасте определяется наибольшая разница в уровнях ОХ здоровых детей и носителей дефектного гена (можно диагностировать СГХС в 88% с 0,1% ложно положительных результатов) [20]. Авторы утверждают, что если эту прослойку населения подвергать генетическому тестированию с последующим каскадным скринингом, то через 30 лет будет известно большинство носителей дефектных генов, после чего можно будет остановить сплошной скрининг и лишь мониторировать затронутые семьи. Каскадный скрининг — обследование родственников пациента первой, второй, а иногда и третьей степени родства на наличие уже выявленной мутации. Данный метод позволяет выявлять СГХС до развития осложнений с возможностью проведения первичной профилактики. Позднее те же авторы обследовали 10095 детей в возрасте 1-2 лет во время плановых посещений поликлиники, в результате СГХС была выявлена у 40 детей (0,4%) и 40 родителей [21]. К преимуществам данного метода можно отнести простоту организации (первичное исследование ОХ крови проводится во время плановой иммунизации); сплошной охват; ранняя диагностика как у детей, так и их родителей. По сообщениям авторов исследования, ни в одном из случаев родители ранее не получали липид-снижающую терапию. Стоит отметить, что в качестве скрининга на СГХС авторами учитывалась лишь тяжелая гиперхолестеринемия (>95 процентиля).
Другой метод — поиск группы риска и целевой скрининг. Пациенты, перенесшие острый коронарный синдром (ОКС) в возрасте менее 55 и 60 лет у мужчин и женщин, соответственно, представляют перспективную когорту для такого скрининга. По данным метаанализа из 22 исследований, включившего 31436 случаев, распространенность СГХС растет со снижением возраста пациентов. Среди пациентов <60 лет распространенность оказалась 7,3%, а при сужении выборки до возраста <45 лет, распространенность диагностики вероятной/определенной СГХС увеличилась до 13,7% [22]. В случае развития ОКС все пациенты с СГХС относятся к очень высокому риску, однако, учитывая молодой возраст, отсутствие известных сопутствующих заболеваний и сердечно-сосудистых событий в анамнезе, при решении вопроса о тактике ведения таких пациентов могут ошибочно отнести к категории низкого риска, что приведет к удлинению сроков проведения внутрисосудистого вмешательства и ухудшению прогноза в случае развития ИМ [23].
Диагностика
Хотя золотым стандартом верификации наследственно обусловленного заболевания является генетический метод, он не всегда доступен. По этой причине были разработаны несколько вариантов клинических критериев диагностики СГХС. Среди них наиболее известные британские (SB — Simon Broome Registry) [24], голландские (DLCN — Dutch Lipid Clinic Network) [25], американские (MEDPED — Make Early Diagnosis to Prevent Early Deaths) [26] и японские (JAS — Japanese Atherosclerotic Society) [27]. Все шкалы оценивают семейный анамнез и уровень ЛНП пациента, кроме того, японские критерии оценивают толщину Ахиллова сухожилия, а SB и DLCN — наличие генетической мутации. DLCN также учитывают анамнез сердечно-сосудистых событий пациента и являются самыми детализированными в оценке вероятности СГХС. Во время прямого сравнения клинических критериев и генетического тестирования в качестве метода диагностики СГХС у 103 пациентов ?65 лет с ОКС были получены следующие результаты: распространенность генетически подтвержденной СГХС составила 8,7%; DLC и SB критерии не диагностировали генетически подтвержденную СГХС в 4 и 3 случаях, соответственно [28]. Причина низкой чувствительности метода может быть связана с широким применением статинов среди населения и искусственно заниженными значениями ЛНП. Доступность пластических операций позволяет многим пациентам избавляться от непривлекательных ксантом и ксантелазм [29], наличие которых увеличивает клиническое подозрение. Таким образом, представленные критерии занимают важное место в диагностике СГХС, однако не могут полностью заменить генетический метод. Необходимость генетического исследования возникает у пациентов с “определенной” СГХС и отсутствием эффекта от высокодозной липид-снижающей терапии. Некоторые мутации, например, гомозиготные нулевые мутации гена LDLR, требуют проведения липидного афереза или трансплантации печени, т.к. пациенты нечувствительны к статинам и ингибиторам PSCK9 [30-32]. Может наблюдаться схожесть клинических проявлений СГХС и других наследственных дислипидемий, как наследственные гипертриглицеридемии, ситостеролемия, дисбеталипопротеимения и др. [32][33]. Таким пациентам также часто требуется специфическая терапия, отличная от стандартного лечения СГХС.
Masana L, et al. (2019) предлагают пересмотреть классификацию СГХС. Авторы призывают считать СГХС синдромом, т.е. сочетанием гиперхолестеринемии, семейного анамнеза и высокого сердечнососудистого риска вследствие различных причин. Синдром СГХС включает как клинические, так и генетические варианты: гетерозиготная СГХС; гомозиготная СГХС; полигенная СГХС; СГХС, комбинированная с гипертриглицеридемией [34]. К гетерозиготной СГХС следует относить также носителей мутаций генов апопротеина Е, адаптерного белка преобразования сигнала семейства 1 (STAP1), пататин-подобного фосфолипазосодержащего домена семейства 5 (PNPLA5) [2]. К гомозиготной форме, кроме гомозигот по генам LDLR, ApoB или PCSK9, также предлагается относить компаудных гетерозигот (оба аллеля одного гена имеют разные мутации), комбинированных гетерозигот (два гена имеют разные мутации) и аутосомно-рецессивную форму гиперхолестеринемии при мутации гена адаптерного белка 1 LDLR (LDLRAP1). Полигенную СГХС авторы предлагают диагностировать при соответствии известным критериям и отсутствии мутаций вышеупомянутых генов [34]. К полигенной СГХС можно отнести от 10 до 50% пациентов по данным различных работ [3][35]. Хотя риск таких пациентов может быть ниже, чем при известных моногенных мутациях, они также нуждаются в активной профилактике и мониторинге с молодого возраста.
Профилактика
Согласно рекомендациям по дислипидемиям 2019г Европейского общества кардиологов, пациенты с СГХС в отсутствии заболеваний атеросклеротического генеза относятся к группе высокого риска развития сердечно-сосудистых осложнении? с целевым значением ЛНП <1,8 ммоль/л, а при их наличии — к группе очень высокого риска, при котором требуется достигать уровня <1,4 ммоль/л или снижения ЛНП на ?50% [6]. Позиция Российского кардиологического общества в рекомендациях по ведению пациентов с семейной гиперхолестеринемией следующая: целевой уровень ЛНП у взрослых пациентов с СГХС долен быть <2,5 ммоль/л, при наличии сердечно-сосудистых заболеваний в анамнезе уровень ЛНП <1,5 ммоль/л, для детей младше 10 лет целевой уровень <4 ммоль/л, а по достижении 10 лет — <3,5 ммоль/л [36]. Рекомендуется начинать липид-снижающую терапию в возрасте 8-10 лет при неэффективности немедикаментозных методов в течение 3-6 мес. [6][36][37]. В пользу первичной профилактики у детей говорят результаты следующего исследования. Авторы взяли когорту из 214 пациентов с СГХС в возрасте 31,7±3,2 лет (98% имели генетическое подтверждение), которые в детском возрасте участвовали в двухлетнем исследовании применения правастатина, измерили уровень ЛНП, оценили анамнез по сердечно-сосудистым событиям/смерти за 18 (15-21) лет. Кроме того, 156 родителей, страдающих СГХС, также были исследованы на предмет неблагоприятных событий. Были получены следующие результаты: 79% пациентов получали липид-снижающую терапию; среди тех, кто прекратил прием, лишь в четырех случаях были отмечены побочные эффекты; не отмечено случаев рабдомиолиза или других тяжелых состояний; среди тех, кто продолжил прием медикаментов, не было замечено повышения уровней печеночных ферментов или креатинфосфокиназы. Группой контроля выступали родные братья и сестры пациентов (n=95), не страдающие СГХС [38]. Так, была продемонстрирована безопасность длительного приема статинов у детей. Уровень ЛНП снизился на 32% по сравнению с первоначальным. При этом ИМ, стенокардия, поражение периферических артерий, инсульт, реваскуляризация миокарда или сердечно-сосудистая смерть произошли в 1% и 0%, соответственно, у пациентов, в 26% и 7%, соответственно, у их родителей, которым терапия была недоступна в раннем возрасте в связи с тем, что статины были открыты только в 1988г, когда большинство родителей пациентов было старше 30 лет [38].
Наследственность играет значимую, но не решающую роль. Регистр “CASCADE-FH”, включающий >1900 пациентов с СГХС, показал, что такие факторы риска, как возраст, мужской пол, артериальная гипертензия, сахарный диабет, ожирение и курение, приводят к повышению частоты сердечно-сосудистых событий у лиц с СГХС [39], демонстрируя важность комплексного подхода в ведении таких пациентов. Схожие данные получены Akioyamen LE, et al. (2019) в ходе метаанализа [40]. Заключение Продолжительность жизни пациента с СГХС напрямую зависит от эффективности как первичной, так и вторичной профилактики сердечно-сосудистых событий; которые возможны при соблюдении ряда условий:
- скрининг пациентов с ранними сердечно-сосудистыми событиями;
- маршрутизация пациентов с высоким уровнем подозрения СГХС в липидные центры;
- доступность генетического анализа, использование клинических критериев в случае невозможности проведения генетического исследования;
- повышение настороженности врачей в отношении СГХС и проведение каскадного скрининга для более эффективной профилактики.
Ведение регистров позволяет не только структурировать уже имеющуюся информацию, но и получать сведения по влиянию дополнительных факторов риска на течение заболевания, эффективности проводимой терапии и прогнозу для жизни у пациентов с уже известными генетическими мутациями. Будущие исследования помогут определить оптимальный метод раннего скрининга на СГХС, а ежегодный рост распространенности заболевания будет свидетельствовать в пользу лучшей диагностики, а значит контроля над ситуацией. Авторы не касались медикаментозной профилактики сердечно-сосудистых событий, однако клинические исследования в данной области также позволят в будущем значимо улучшить прогноз пациентов с СГХС.
Пациенты в представленных клинических случаях дали свое согласие на публикацию информации без раскрытия имен.
Отношения и деятельность: все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье.