Нестандартные подходы к лечению фибрилляции предсердий — взгляд за рамки клинических рекомендаций
Аннотация
В статье представлены литературные данные и результаты собственного клинического опыта, свидетельствующие о том, что фибрилляция предсердий (ФП), являясь стереотипным клиническим проявлением патологического фиброзирования миокарда, может иметь в своей основе различные, не всегда очевидные пусковые факторы. Приведены морфологическая и клиническая классификации фиброза. Обоснована полиэтиологичность миокардиального фиброза, описаны механизмы его патогенеза, реализующиеся через гипоксическое повреждение митохондрий кардиомиоцитов. Сделан акцент на некоторых неочевидных причинах митохондриальной дисфункции, которые широко распространены в популяции и могут быть причиной резистентности ФП к антиаритмическому и интервенционному лечению. В абсолютном большинстве случаев эти этиологические факторы остаются не выявленными, т.к. не включены в алгоритмы текущих рекомендаций. Приведены собственные клинические данные нейромодуляционного лечения пожилых коморбидных пациентов с ФП и сердечной недостаточностью, сформулирована гипотеза возможной регенерации миокарда, основанная на стимуляции дифференцировки эндогенных миокардиальных стволовых клеток. Составлен алгоритм, которым, по мнению автора, целесообразно дополнить стандартное обследование пациентов с ФП для улучшения долгосрочной эффективности лечения.
Традиционное лечение фибрилляции предсердий (ФП) сосредоточено на контроле ритма или частоты сердечных сокращений, профилактике тромбоэмболических осложнений и модификации факторов риска, ассоциированных с сердечно-сосудистыми заболеваниями (ССЗ). Однако растущее понимание патофизиологии ФП смещает фокус внимания в сторону патогенетической терапии, направленной на устранение глубинных причин, инициирующих и поддерживающих аритмогенный субстрат, основой существования и прогрессирования которого являются профиброгенные процессы в миокарде предсердий. Среди таких причин — полиэтиологические метаболические дисфункции кардиомиоцитов (КМЦ), латентные воспалительные процессы и нарушения нейрогормональной регуляции, которые часто взаимосвязаны и остаются за рамками стандартных алгоритмов, являясь, между тем, дополнительным ключом к действительно персонализированной этиотропной и патогенетической терапии ФП.
I. Особенности строения и функционирования предсердных КМЦ
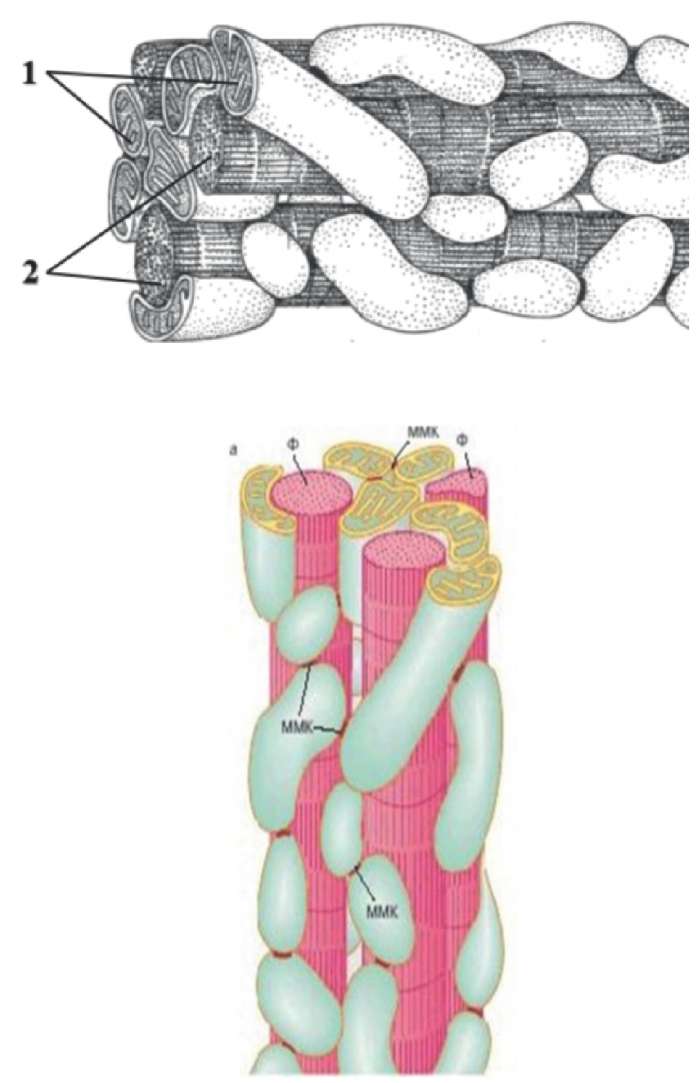
Фундаментальным отличием миокарда от других органов и систем является не только способность к сокращению, но и огромное количество митохондрий, содержащихся в КМЦ — до 30-35% объема клетки (рис. 1), что обусловлено высокой потребностью в АТФ в связи с необходимостью постоянного сокращения. Это делает сердце в целом в высшей степени зависимым от кислорода и сердечные клетки крайне уязвимыми к гипоксии, даже более, чем клетки головного мозга.

Рис. 1. Строение КМЦ.
Примечание: 1. митохондрии; 2. миофибриллы.
Сокращения: Ф — миофибриллы, ММК — межмитохондриальные контакты.
1. Особенности строения предсердного миокарда
Предсердные КМЦ имеют уникальные черты строения по сравнению с желудочковыми:
— Специфический профиль ионных каналов, обеспечивающих меньшую продолжительность потенциала действия. Например, в предсердных КМЦ выражены ультрабыстрый калиевый ток (IKur), который практически отсутствует в желудочках, а также другие специфичные токи (IK,ACh), что делает их потенциал действия короче. Эта особенность обеспечивает высокую частоту сокращения предсердий.
— Слабо развитая система Т-трубочек (инвагинации сарколеммы), что определяет особенности кальциевого гомеостаза и способствуют возникновению кальций-зависимой триггерной активности (поздних постдеполяризаций), которая является одним из ключевых механизмов инициации и поддержания ФП. Отсутствие развитой сети Т-трубочек приводит к тому, что инициирование кальциевого трансмембранного тока происходит преимущественно на периферии клетки. Это создает пространственно-временную гетерогенность в цитозольной концентрации Ca²⁺, что является одним из ключевых факторов, способствующих возникновению аритмий. Кроме того, современная модель предполагает, что в предсердных КМЦ ключевую роль играют не инвагинации сарколеммы, а специфические так называемые «нановилы», где кальциевые каналы L-типа (медленные) тесно сопряжены с кластерами рианодиновых рецепторов (RyR2) на мембране саркоплазматического ретикулума. Нарушение этого тонкого сопряжения при патологии (прежде всего при ФП) является драйвером нарушений сократимости и формирования электрической нестабильности.
— Наличие секреторных гранул: КМЦ предсердий выполняют эндокринную функцию, выделяя, в ответ на растяжение, предсердный натрийуретический пептид, в меньшей степени — мозговой натрийуретический пептид и другие родственные пептиды и нейротрансмиттеры. Существуют исследования, подтверждающие наличие в предсердиях клеток, способных к локальному синтезу катехоламинов (норадреналина) — собственные адренергические клетки сердца. Вся эта система играет ключевую роль в регуляции вегетативного баланса, кровяного давления и объема крови. Современные исследования показывают, что предсердный натрийуретический пептид и прочие эндогенные пептиды, секретируемые предсердными КМЦ, оказывают не только системное действие, но и аутокринное/паракринное влияние на сами КМЦ, регулируя их метаболизм и гипертрофический ответ, а также контролируя локальное реактивное воспаление.
— Более высокая метаболическая пластичность. В норме предсердные КМЦ в большей степени, чем желудочковые, полагаются на окисление глюкозы (гликолиз), но при стрессовых воздействиях (нагрузка давлением или объемом, ФП и пр.) их метаболизм нарушается, что способствует развитию диастолической дисфункции и быстрому аритмогенному ремоделированию. Предсердный миокард содержит больше липидов, чем миокард желудочков и поэтому более подвержен структурной аритмогенной трансформации.
Всё это в совокупности напрямую влияет на электрофизиологические свойства и делает предсердные КМЦ функционально способными к более быстрому проведению импульса, обуславливает большую зависимость от электролитного дисбаланса, преимущественно кальциевого, и, следовательно, определяет большую уязвимость к возникновению аритмий, в особенности ФП.
Предрасполагающим анатомо-гистологическим фактором к формированию макро- и микрореентри, очагов фокусной активности является то, что предсердия имеют более сложную, чем желудочки трёхмерную структуру. Она включает межпредсердные соединения, ограниченные пучком Бахмана, межжелудочковой перегородкой и коронарным синусом; гребенчатые мышцы, конечный гребень и волокна, окружающие коронарный синус в правом предсердии; а также легочные вены со сложной ориентацией волокон вокруг них в левом предсердии (ЛП).
Эти структурные особенности имеют важные потенциальные последствия для патофизиологии предсердий и лечения предсердных аритмий [1].
2. Особенности иннервации предсердий
Особенностью вегетативной иннервации является существование собственной сложноорганизованной «внутренней нервной системы сердца», состоящей из сплетения постганглионарных нервных окончаний и интракардиальных ганглиев. В отличие от желудочков, плотность холинергических (парасимпатических) окончаний в миокарде предсердий в норме значительно выше, чем адренергических (симпатических). Постганглионарные нейроны парасимпатической нервной системы расположены непосредственно в эпикарде или в толще миокарда предсердий, и формируют сложные интракардиальные ганглионарные сплетения, расположенные преимущественно внутри жировых подушек. Реализация факторов риска ССЗ приводит к ремоделированию предсердий и формированию симпатической гипериннервации. Патологическая катехоламиновая гиперстимуляция укорачивает эффективный рефрактерный период предсердных КМЦ и продолжительность потенциала действия, усиливает дисперсию деполяризации и способствует возникновению поздних постдеполяризаций, создавая условия для возникновения и поддержания предсердных аритмий. Длительная хроническая гиперсимпатикотония приводит к вазоконстрикции, повышению системного артериального давления и тахикардии, что увеличивает потребность миокарда в кислороде и может вызывать относительную ишемию предсердий даже при отсутствии коронарного атеросклероза. Длительная субклиническая ишемия и прямое цитотоксическое действие катехоламинов способствуют активации фибробластов. Увеличение доли фиброза в миокарде предсердий — ключевой элемент структурного ремоделирования, который нарушает нормальное проведение импульса и изолирует группы КМЦ, способствуя персистированию ФП. Таким образом, дисбаланс автономной регуляции сердца замыкает «порочный круг» патогенеза ФП [2].
3. Особенности кровоснабжения предсердий
Особенность кровоснабжения предсердий заключается в его прямой связи с проводящей системой сердца, что делает нарушения в этом бассейне высокоаритмогенными. Основным источником кровоснабжения правого предсердия являются тонкие ветви правой коронарной артерии (ПКА): артерия синусового узла (в 55-65% случаев отходит от ПКА, обеспечивая кровью не только синусовый узел, но и большую часть стенки правого предсердия; в 35-45% случаев артерия синусового узла отходит от огибающей ветви левой коронарной артерии, тогда она также питает часть ЛП); правая краевая ветвь (иногда — артерия острого края) — питает латеральную стенку; мелкие предсердные ветви от начального отдела ПКА. ЛП кровоснабжается в основном через тонкие предсердные ветви огибающей артерии, включая артерию атриовентрикулярного узла и косую артерию ЛП, идущую по задней поверхности ЛП. Межпредсердная перегородка кровоснабжается от ветви предсердно-желудочкового узла (от ПКА или огибающей) и мелких ветвей от обеих коронарных артерий. Предсердия имеют развитую сеть межкоронарных анастомозов, что может обеспечивать коллатеральный кровоток при ишемии одной из ведущих артерий. Основной кровоток идет через интрамуральную микроциркуляторную сеть, функционирование которой нарушается при диастолической дисфункции предсердий. При длительной перегрузке предсердий объемом или давлением (клапанные пороки сердца, артериальная и легочная гипертензии) происходит разрежение капиллярной сети (редукция капиллярной плотности) в миокарде предсердий, что напрямую ограничивает доставку кислорода. Микрососуды предсердий высокочувствительны к эндотелиальной дисфункции и склонны к спазму под воздействием оксидативного стресса, провоспалительных цитокинов и нейротрансмиттеров симпатической нервной системы (СНС) [3].
Таким образом, нормальное кровоснабжение миокарда предсердий и проводящей системы критично для удержания синусового ритма, а атеросклеротическое поражение предсердных артерий в большинстве случаев недоступно для хирургической или интервенционной коррекции.
II. Фиброз как патогенетический субстрат ФП
Фиброз миокарда — это сложный и многогранный патологический процесс замещения функциональных КМЦ элементами межклеточного матрикса. Он является универсальным ответом сердца на повреждение и лежит в основе многих ССЗ, замыкая «порочный круг» в патогенезе.
В здоровом миокарде межклеточный матрикс составляет не более 20% и состоит из коллагеновых волокон преимущественно I и III типов, эластина, фибронектина, неструктурных гликопротеинов и гликозаминогликанов. Баланс между синтезом и распадом коллагена обеспечивает нормальную структуру и функцию сердечной мышцы. Деградация избытка соединительнотканных веществ происходит с помощью металлопротеиназ.
Патологический фиброз, т.е. избыточный синтез коллагенов в миокарде предсердий, развивается вследствие активации миофибробластов при механическом повреждении, растяжении, хроническом воспалении, иммунных реакциях, нейрогуморальных и метаболических нарушениях, формируя так называемую атриопатию или предсердную миопатию, на фоне которой становится возможной инициация ФП [4].
По гистологической структуре выделяют 3 основных варианта фиброза [5] (рис. 2):
- Репаративный или заместительный (очаговый) фиброз — возникает в ответ на острое состояние, чаще всего при некрозе сердечной мышцы на месте утраченных КМЦ, и является по сути адаптивной, органосохраняющей реакцией на повреждение.
- Интерстициальный или реактивный (диффузный или очаговый) фиброз — увеличение объема межклеточного матрикса за счет увеличения синтеза коллагена при сохранных КМЦ, возникает как патологическая реакция в ответ на хроническое неблагоприятное воздействие.
- Периваскулярный (чаще — диффузный) фиброз — отложение соединительной ткани вокруг компонентов микрососудистого русла (часто присоединяется к поздним стадиям репаративного или интерстициального фиброза).

Рис. 2. Патологический фиброз миокарда.
Сокращения: ВСС — внезапная сердечная смерть, ХСН — хроническая сердечная недостаточность.
Диффузный интерстициальный фиброз является общепризнанным краеугольным звеном в формировании аритмогенного субстрата для возникновения и поддержания ФП. Предсердия значительно более склонны к развитию диффузного интерстициального фиброза, чем желудочки. Эта разница обусловлена комплексом анатомических, гемодинамических, молекулярных и биохимических факторов [6].
Миокард предсердий значительно тоньше, чем у желудочков. В результате одно и то же повреждающее воздействие, например воспаление или растяжение (перегрузка объемом или давлением), оказывает на более тонкую стенку предсердия относительно большее влияние.
Анатомия предсердий сложнее, чем желудочков и, в связи с обилием структур физиологической соединительной ткани, контактирующей с миокардиальной (муфты легочных вен, ушки, устье коронарного синуса, места впадения системных вен), создаются предпосылки для формирования зон замедленного проведения. Кроме того, предсердные КМЦ имеют особенности распределения белков межклеточного взаимодействия, экспрессируя два вида коннексинов 43 и 40, тогда как желудочковые КМЦ экспрессируют исключительно коннексин-43. Неоднородность в распределении коннексина-40 в миокарде предсердий также создает предпосылки для неоднородного проведения электрических импульсов и формирования интерстициального фиброза [4][7].
Кроме того, повышенная чувствительность к нейрогормонам и формирование порочного круга с ФП делают предсердия уникальной и уязвимой мишенью для патологического ремоделирования [7].
Диагностика фиброза миокарда основана на лабораторных данных, визуализационных методиках и гистологических исследованиях, но выявление интерстициального фиброза предсердий, как основного аритмогенного субстрата для ФП, имеет существенные ограничения (табл. 1).
Таблица 1
Клинические методы диагностики интерстициального фиброза миокарда предсердий
|
Методика |
Суть методики |
Преимущества |
Недостатки |
Основное применение |
|
Эхо (strain) |
Оценка деформации миокарда |
Неинвазивно, доступно для популяционного скрининга |
Косвенная оценка, зависит от оператора |
Скрининг, динамическое наблюдение |
|
МРТ (strain) |
Оценка деформации миокарда |
Неинвазивно |
Косвенная оценка, дорого, не везде доступно, требует опыта |
Динамическое наблюдение |
|
МРТ (LGE) |
Визуализация задержки контраста в фиброзной ткани |
«Золотой стандарт» неинвазивной количественной оценки фиброза в миокарде |
Не дифференцирует заместительный и интерстициальный фиброз, не выявляет начальные стадии интерстициального фиброза предсердий |
Прогноз |
|
Лабораторные тесты |
Оценка концентрации в сыворотке крови белков, связанных синтезом и распадом коллагенового матрикса миокарда |
Неинвазивно, доступно для скрининга |
Невысокая чувствительность и специфичность, не является самостоятельным критерием диагностики |
Динамическое наблюдение, возможно, скрининг |
|
Электро-физиологическое исследование |
Оценка амплитуды электрических сигналов с поверхности миокарда |
Прямая оценка субстрата аритмии в реальном времени |
Инвазивно, проводится только в ходе радиочастотной аблации |
Не применима для популяционного скрининга |
|
Биопсия |
Гистологическое исследование ткани |
Прямая и самая точная визуализация |
Высокая инвазивность, точечная, риск осложнений |
Не применима для популяционного скрининга |
Сокращение: МРТ — магнитно-резонансная томография.
На прогрессирование фиброза указывает ряд лабораторных тестов: повышение концентрации С-концевого пептида проколлагена I, N-концевого пропептида коллагена III, тканевого ингибитора матриксной металлопротеиназы и пр. Эти лабораторные маркеры имеют умеренную чувствительность и специфичность, поэтому должны дополняться другими методами диагностики [8].
Разрешающие способности магнитно-резонансной томографии (МРТ) миокарда с контрастированием на современном этапе клинической практики не позволяют визуализировать интерстициальный фиброз предсердий: расчетные показатели Т1 и ECV не применяются в связи с тонкостью миокарда, а LGE не позволяет корректно дифференцировать заместительный и диффузный фиброз в миокарде предсердий [9][10]. Прижизненная биопсия предсердий, являющаяся «золотым стандартом» диагностики любого фиброза, не может быть рутинным методом. Косвенными визуализирующими методиками оценки фиброза являются на сегодня strain-эхо и strain-МРТ [11]. В аритмологической операционной для косвенной оценки фиброза традиционно используется электро-анатомическое вольтажное картирование.
Важна также идентификация клинической стадийности интерстициального фиброза (Шевченко Ю. Л. и др., 2022 [12]):
I — латентная стадия, период бессимптомного течения (ранние изменения соединительной ткани);
II — стадия первичных минимальных проявлений (умеренная степень фиброза с увеличением коллагена I и III типа);
III — стадия диастолической дисфункции (выраженная степень фиброза со значимым преобладанием коллагена I типа);
IV — стадия систолической и диастолической дисфункции (тяжелая степень фиброза);
V — стадия коронарной ангиопатии (крайне тяжелая степень фиброза с вовлечением периферического коронарного русла).
Вероятно, что I, и отчасти II, клинические стадии интерстициального фиброза могут быть обратимы, при условии этиотропного и патогенетического подхода к лечению атриопатии.
III. Основные причины формирования и прогрессирования интерстициального фиброза предсердного миокарда
1. Хроническое субклиническое системное воспаление
Хроническое латентное или «низкоуровневое» системное воспаление представляет собой вялотекущий, клинически малозаметный процесс, характеризующийся постоянным, умеренно повышенным уровнем провоспалительных цитокинов и маркеров воспаления в тканях и плазме крови. В отличие от острого воспаления, которое является защитной реакцией, это состояние незаметно повреждает мембраны клеток, нарушает митохондриальное дыхание и является ключевым патогенетическим звеном в развитии атеросклероза, диабета 2 типа, фиброза миокарда, нейродегенеративных и аутоиммунных заболеваний [13].
Ключевыми причинами поддержания латентного системного воспаления являются:
1.1. Метаболические дисфункции
— Ожирение и висцеральный жир (в т.ч. так называемое «ожирение нормального веса»): адипоциты, особенно висцеральной жировой ткани, продуцируют провоспалительные адипокины (фактор некроза опухоли-α (ФНО-α), интерлейкин (ИЛ)-6, лептин). Макрофаги инфильтрируют гипертрофированную жировую ткань, формируя «коронные структуры» вокруг гибнущих адипоцитов и усиливая воспалительный ответ.
— Инсулинорезистентность: формирует порочный круг — воспаление способствует резистентности к инсулину, а гиперинсулинемия и гипергликемия, в свою очередь, активируют каскад системного воспаления.
— Диета с высоким содержанием насыщенных жиров, простых сахаров, ультрапереработанных продуктов и конечных продуктов гликирования, приводит к дефициту пищевых волокон, полиненасыщенных жирных кислот ω-3, антиоксидантов и полифенолов.
Таким образом, метаболический синдром — это не просто совокупность факторов риска, а системное провоспалительное состояние, запускающее каскад процессов, ведущих к повреждению тканей, в т.ч. миокарда предсердий. Висцеральное ожирение, инсулинорезистентность, артериальная гипертензия и дислипидемия (не редко в совокупности с гиперурикемией) ведут к активации ренин-альдостерон-ангиотензиновой системы. Ангиотензин II является мощным стимулятором пролиферации фибробластов и синтеза коллагена, усиливает окислительный стресс КМЦ. Инсулинорезистентность и гипергликемия приводят к генерации активных форм кислорода (АФК), которые непосредственно повреждают КМЦ и стимулируют фиброгенные сигнальные пути (например, трансформирующий фактор роста-бета1). Адипоциты висцеральной жировой ткани секретируют провоспалительные цитокины: ФНО-α, ИЛ-6, ИЛ-1β. Эти цитокины попадают в миокард и активируют резидентные макрофаги и фибробласты. Избыток свободных жирных кислот и глюкозы нарушает метаболизм КМЦ, запуская апоптоз. Наконец, белки острой фазы, повышенные в той или иной степени при метаболическом синдроме, оказывают прямое цитотоксическое действие на КМЦ и модулируют активность ионных каналов, снижая реполяризационный резерв мембран, а также запускают паракринную активность собственных адренергических клеток сердца, секретирующих норэпинефрин в межклеточное пространство, что приводит к гиперстимуляции β1-адренорецепторов КМЦ и формированию патологической гиперсимпатикотонии.
Хроническое латентное воспаление диагностируется не симптоматически, а по лабораторным маркерам: повышение высокочувствительного С-реактивного белка — «золотой стандарт» оценки системного воспаления низкой степени; увеличение уровня провоспалительных цитокинов: ИЛ-6, ИЛ-1β, ФНО-α и пр., реже — повышение скорости оседания эритроцитов, фибриногена, ферритина. Выявление латентной фазы системного воспаления критически важно для превентивной кардиологии в целом, для выявления неблагоприятных вариантов течения заболевания и отслеживания динамики на фоне проводимой терапии. Поэтому лабораторная диагностика низкоуровневого системного воспаления, наряду с маркерами преддиабета (инсулин, гликированный гемоглобин), должны быть включены в рутинные алгоритмы обследования пациентов с ФП [13-15].
1.2. Хронические инфекции и хроническое аутоиммунное воспаление
— Персистирующие кардиотропные вирусы (цитомегаловирус, вирус Эпштейна-Барр, герпесвирусы) постоянно стимулируют иммунную систему, истощая ресурсы Т-клеток и поддерживая хроническую антигенную напряженность. Хронические очаги бактериальной инфекции (например, пародонтит, вызванный Porphyromonas gingivalis) становятся постоянными источниками провоспалительных медиаторов, обладающих цитотоксическими свойствами [16].
— Латентное аутоиммунное воспаление представляет собой доклиническую или субклиническую стадию аутоиммунного процесса, при которой происходит специфическая активация иммунной системы против собственных антигенов без развернутой симптоматики органного поражения. Это состояние является важным компонентом и часто пусковым механизмом системного низкоуровневого воспаления, создавая патологическую основу для развития манифестных аутоиммунных заболеваний и сердечно-сосудистых осложнений [17].
Латентное аутоиммунное воспаление поддерживает системный воспалительный процесс несколькими путями:
1) Хроническая антигенная стимуляция, когда персистирующие аутоантигены постоянно поддерживают активность иммунной системы, приводя к хронической активации моноцитов/макрофагов, дендритных клеток и лимфоцитов.
2) Секреция провоспалительных цитокинов: ФНО-α, ИЛ-1, 6 и пр., которые действуют системно, нарушая функцию эндотелия, способствуя инсулинорезистентности и активируя острофазовый ответ в печени.
Системное воспаление, индуцированное аутоиммунным процессом, усугубляет инсулинорезистентность и дислипидемию. В свою очередь, метаболический синдром создает провоспалительную среду, которая еще больше подстегивает аутореактивность [17].
Известно, что хроническое воспаление активирует ось гипоталамус — гипофиз –надпочечники, формируя патологическую гиперсимпатикотонию. Повышение уровня кортизола и катехоламинов (гормонов и нейротрансмиттеров СНС), которые в хроническом режиме модулируют иммунный ответ в сторону провоспалительного профиля, поддерживает системное воспаление [18-20].
2. Латентный миокардит — скрытый фактор формирования и прогрессирования интерстициального фиброза предсердий
Хронический латентный миокардит, который может годами протекать малосимптомно, не приводя к значимому увеличению объемов сердца, но вызывая прогрессирующий интерстициальный фиброз, является одним из самых распространенных и недооцененных факторов запуска и поддержания профибротических и проаритмогенных процессов в миокарде предсердий.
Ряд аутопсийных и прижизненных гистологических исследований у пациентов с ФП демонстрируют высокую встречаемость лимфоцитарной инфильтрации предсердий, соответствующую критериям диагностики миокардита. Причем важно, что в абсолютном большинстве случаев такие латентные проаримогенные формы миокардита без дилатации камер сердца протекают по аутоиммунному и реактивному варианту, являясь органным проявлением системного воспаления и/или исходом в аутоиммунную фазу вирусного поражения миокарда, т.к. в биоптатах крайне редко обнаруживаются вирусные, а тем более бактериальные, антигены [21].
В основе механизмов формирования и поддержания латентного аутоиммунного воспаления в миокарде лежат два механизма: 1) перекрестная реактивность, когда иммунный ответ на внешние кардиотропные факторы (инфекционные агенты, ксенобиотики и пр.) может переключиться на КМЦ; и 2) обнаружение иммунной системой структур, в норме не доступных для распознавания, и выработка антительного ответа (например антитела к ядрам КМЦ, обнажившихся в результате цитолиза). Циркулирующие иммунные комплексы (комплексы аутоантител с аутоантигенами), даже в небольших количествах, могут откладываться в тканях (миокардиальный интерстиций, сосудистый эндотелий), активировать систему комплемента и поддерживать системное и локальное воспаление. И, конечно, процесс хронического воспаления в миокарде приводит к формированию диффузного интерстициального фиброза, запуская каскад аритмогенеза [22].
Диагностика латентного миокардита основывается на выявлении специфических антител к структурам КМЦ и проведению МРТ миокарда с контрастированием. Выявление МРТ-признаков отека миокарда желудочков и феномена ECV и LGE в сочетании с мерцательной аритмией, часто при отсутствии острофазовых лабораторных показателей позволяет с высокой степенью вероятности диагностировать вовлечение в воспалительный процесс миокарда предсердий [22-24].
3. Хроническая гипоксия
В отличие от острой ишемии, которая быстро приводит к некрозу, хроническая гипоксия КМЦ запускает сложные адаптационные и дезадаптационные процессы, лежащие в основе профибротических процессов в миокарде. В основе патогенеза интерстициального предсердного фиброза чаще всего лежит сочетание различных форм гипоксии [25]. Гипоксемическая гипоксия — снижение парциального давления кислорода в артериальной крови (нарушение оксигенации крови) у кардиологических пациентов чаще всего наблюдается при альвеолярной гиповентиляции (ожирение, хроническая обструктивная болезнь легких). Гемическая гипоксия, обусловленная нарушением функционирования гемоглобина, возникает в большинстве клинических случаев на фоне хронической железодефицитной анемии и латентного железодефицита. Ишемическая гипоксия развивается при атеросклеротическом поражении ветвей коронарных артерий, кровоснабжающих предсердия, или на фоне диастолической дисфункции миокарда предсердий вследствие редукции предсердной капиллярной сети. Тканевая гипоксия наблюдается при нарушении утилизации кислорода тканью, что может быть следствием нарушения синтеза ферментов и/или снижения их активности, а также разобщения процессов окисления и фосфорилирования в митохондриях на фоне метаболических дисфункций, хронического низкоуровневого системного воспаления, латентных миокардитов и хронических интоксикаций.
Любые клинически значимые гипоксические состояния ведут прежде всего к повреждению митохондрий предсердных КМЦ, т.к. по сравнению с желудочковыми, предсердные КМЦ имеют меньший объем митохондрий (20-25% vs 30-35% в желудочках), а клетки проводящей системы предсердий (особенно синусового узла) имеют высокий уровень базального метаболизма и их митохондрии более уязвимы к энергетическому дефициту, что проявляется дисфункцией синусового узла, блокадами внутрипредсердного проведения и предсердными аритмиями. Митохондрии выступают здесь как детектор энергетического дефицита, генератор оксидативного стресса, регулятор кальциевого гомеостаза и триггер апоптоза КМЦ. Гибель КМЦ через апоптоз, запускаемый митохондриями, стимулирует заместительный фиброз предсердий и реактивный очаговый миокардит, приводящий к интерстициальному фиброзированию [25]. Таким образом, именно митохондриальная дисфункция, возникшая в ответ на хроническую гипоксию, является центральным звеном предсердного аритмогенеза. Поэтому восстановление митохондриальной функции КМЦ должно лежать в основе долгосрочной стратегии профилактики и лечения ФП.
3.1. Хронические железодефицитные состояния
Железодефицит — глобальная проблема здравоохранения, затрагивающая, предположительно, >2 млрд. человек во всем мире. К сожалению, часто мы обращаем внимание лишь на его конечную стадию — железодефицитную анемию, когда запасы железа исчерпаны и страдает синтез гемоглобина. Однако задолго до анемии развивается латентный дефицит железа — состояние, при котором запасы железа уже истощены, но уровень гемоглобина и эритроцитов еще остается в пределах нормы. Именно на этой стадии важно выявить и скорректировать проблему, предотвратив серьезные последствия. По данным Всемирной организации здравоохранения (ВОЗ), дефицит железа занимает первое место среди причин 38 наиболее распространенных заболеваний человека1, и встречается, по данным различных исследований не менее, чем у 30% общей популяции, достигая 40-60% у женщин репродуктивного возраста, профессиональных спортсменов и пожилых. Согласно данным Osendarp S, et al. (2010) при частоте железодефицитной анемии 20% скрытый железодефицит существует у 50% населения в популяции, а при частоте анемии 40% и выше вся популяция имеет различные степени истощения депонированного железа [26].
Вероятно, латентный железодефицит является основным по распространенности неучтенным фактором митохондриальной дисфункции предсердных КМЦ. Хорошо известно, что помимо непосредственного переноса кислорода через митохондриальную мембрану гемовым железом (рис. 3), его критическая роль заключается в формировании железо-серных кластеров (Fe-S кластеры), которые являются жизненно важными кофакторами для синтеза множества митохондриальных белков. Поэтому митохондриальная дисфункция при латентном дефиците железа развивается задолго до снижения гемоглобина.

Рис. 3. Реакция митохондриального окисления.
Сокращение: АТФ — аденозинтрифосфат.
Собственно хроническая железодефицитная анемия — конечная стадия железодефицита, является причиной 80% всех анемий и выявляется не менее чем у 25-30% населения планеты1.
Основным маркером латентного железодефицита является ферритин сыворотки. Его уровень ниже 30 мкг/л свидетельствует о дефиците железа. Важно, что ферритин является белком острой фазы воспаления, поэтому при наличии воспалительного процесса его уровень может быть ложно повышен. Коэффициент насыщения трансферрина железом (сатурация трансферрина) рассчитывается как отношение сывороточного железа к общей железосвязывающей способности сыворотки, и при латентном железодефиците обычно снижен (<20%). Сывороточное железо — лабильный показатель, зависит от многих факторов (времени суток, приема пищи), поэтому изолировано не используется для диагностики латентного дефицита железа [27].
В недавнем исследовании прогностического значения дефицита железа у пациентов с ФП, с хронической сердечной недостаточностью (ХСН) и без нее, было убедительно показано, что латентный железодефицит, определяемый по уровню сатурации трансферрина TSAT <20%, был связан с более высокой общей смертностью и смертностью от ССЗ у пациентов с ФП, независимо от наличия сопутствующей сердечной недостаточности (СН) [28].
Диагностика латентного железодефицита основана на расчете логарифмического соотношения ферритина и растворимых рецепторов трансферрина:
депо Fe (мг) = 1250 × (log(sTfR/Ферритин) — 2,8229),
где sTfR (soluble Transferrin Receptor) — концентрация растворимых рецепторов трансферрина в сыворотке крови (в мг/л). Ферритин — концентрация ферритина в сыворотке (в мкг/л). Логарифм — обычно используется десятичный логарифм (log10).
Коэффициенты (2,8229 и 0,1207) — получены эмпирически в ходе регрессионного анализа.
Эта формула является «золотым стандартом» в гематологии и позволяет оценить общие запасы железа в организме (включая костный мозг, печень, селезенку) в миллиграммах, и дает возможность точно рассчитать дозировку препаратов железа для конкретного пациента, что полностью исключает риски ятрогенного гемосидероза. Важно, что для назначения препаратов железа в случае выявления истощения его депо, уровень гемоглобина и эритроцитов не имеет значения. Однако необходимо помнить, что приведенная формула может быть корректной при условии отсутствия воспалительного повышения ферритина, поэтому её использование возможно только при нормальном уровне высокочувствительного С-реактивного белка [29].
3.2. Хронический йододефицит
По данным ВОЗ, >30% населения планеты живет в условиях природного йодного дефицита, а патологические состояния, обусловленные йододефицитом, занимают первое место в мире по распространенности среди неинфекционных болезней. В Российской Федерации >70% населения проживает в районах, эндемичных по йододефициту2.
Традиционно йод рассматривается исключительно как обязательный микроэлемент для синтеза тиреоидных гормонов. Однако накопленные за последние десятилетия данные убедительно демонстрируют, что йод (в форме йодид-аниона, I⁻ и молекулярного йода, I2) обладает собственными, независимыми от функции щитовидной железы, биологическими эффектами. Эти эффекты реализуются в различных тканях и органах, включая молочную железу, слюнные железы, желудок, кожу и, что особенно важно, клетки иммунной системы3.
Хроническое системное воспаление низкой степени активности, как уже говорилось, является краеугольным камнем не только формирования атриопатии, но и ключевым аспектом патогенеза эндотелиальной дисфункции, атеросклероза, метаболического синдрома и СН.
В настоящее время известно, что йод улучшает функцию эндотелия, не только за счет нормализации уровня тиреоидных гормонов, но и за счет прямого снижения экспрессии молекул адгезии (VCAM-1, ICAM-1) и увеличения продукции эндотелиального оксида азота (NO), препятствуя таким образом атерогенезу3.
Доказано, что йод (в форме йодид-аниона) ингибирует активацию фактора транскрипции NF-κB, который является главным индуктором экспрессии провоспалительных цитокинов (ФНО-α, ИЛ-1β, ИЛ-6). Предварительные данные свидетельствуют также, что йод может модулировать сборку инфламмасомы NLRP3 (внутриклеточный белковый комплекс, отвечающий за выработку ИЛ), ограничивая развитие реактивного воспаления в органах и тканях. Известно также, что йодид-анион подавляет поляризацию макрофагов в провоспалительный фенотип (M1) и стимулирует их переход в противовоспалительный, репаративный фенотип (M2), что способствует купированию тканевого воспаления [30]. В присутствии пероксидаз йодид-анион (I⁻) окисляется, выступая как «ловушка» для АФК и защищает клетки от окислительного стресса. КМЦ могут быть пассивными бенефициарами этого системного или локального эффекта.
Хорошо изучены метаболические эффекты йода, которые указывают, что отсутствие йододефицита ассоциируется с лучшим гликемическим контролем, снижением инсулинорезистентности и более благоприятным липидным профилем, что частично объясняется подавлением воспаления в жировой ткани и печени и дает дополнительные возможности в лечении метаболического синдрома. Наконец, известно, что дефицит йода косвенно связан с железодефицитом, т.к. йод через гормоны щитовидной железы участвует в регуляции обмена железа, а его недостаток влияет на синтез гемоглобина и метаболизм железосодержащих белков (Fe-S — кластеров), что может ухудшить усвоение железа в тонком кишечнике [31][32].
Однако следует помнить, что резкий избыток йода может, напротив, выступать как провоспалительный стимул и триггер для развития аутоиммунного тиреоидита у генетически предрасположенных лиц, индуцируя системный окислительный стресс, апоптоз и неоантигены в щитовидной железе.
Проаритмогенные эффекты гипо- и гиперфункции щитовидной железы хорошо известны клиницистам [33]. Также известно, что абсолютное большинство случаев гипотиреоза обусловлено хроническим йододефицитом. Но латентная дисфункция щитовидной железы, когда тиреотропный гормон (ТТГ) находится в пределах референсных значений, а синтез гормонов изменен, очень часто остается нераспознанной, т.к. в большинстве случаев кардиологи и даже эндокринологи всё ещё ограничиваются определением только уровня ТТГ в лабораторных исследованиях.
Несколько метаанализов, опубликованных в последнее десятилетие, показывают, что как субклинический гипертиреоз, так и субклинический гипотиреоз связаны с повышенным риском возникновения ФП [34-36]. Важно также отметить, что снижение активной формы трийодтиронина (Т3) возможно при приеме бета-адреноблокаторов (β-АБ), т.к. он, ингибирует действие фермента дейодиназы, ответственного за периферическое превращение неактивного тироксина (Т4) в активный Т3. Это приводит к снижению уровня активного гормона Т3 в крови и может вызывать симптомы, схожие с гипотиреозом, особенно при длительном и в высоких дозах применении β-АБ (ТТГ при этом остается нормальным). Кроме того, β-АБ могут нивелировать клинические симптомы гипертиреоза, т.к. быстро снижают плазменную концентрацию активного Т3, приводя к ложнонормальному ТТГ при повышенном Т4 как общем, так и свободном [37]. Поэтому в повседневной клинической практике аритмолога чрезвычайно важно не ограничиваться только определением уровня ТТГ, но обязательно анализировать значения активных и неактивных тиреоидных гормонов, для диагностики субклинических дисфункций щитовидной железы (Т3 общего и свободного, Т4 общего и свободного + антитела к тиреоглобулину и тиреопероксидазе).
Устранение йододефицита в популяции может быть рассмотрено не только как профилактика заболеваний щитовидной железы, но и как мера, способствующая снижению системного провоспалительного фона, что особенно важно для групп риска по ССЗ и метаболическим заболеваниям.
3.3. Хроническая интоксикация тяжелыми металлами — неучтенная причина митохондриальной дисфункции и резистентности к антиаритмической терапии и интервенционному лечению
Хроническая интоксикация тяжелыми металлами представляет собой скрытую и масштабную угрозу для здоровья человека. В отличие от острых отравлений, хроническое воздействие низких доз часто протекает субклинически, нанося кумулятивный ущерб нервной, сердечно-сосудистой, выделительной и репродуктивной системам. Оценка распространенности является сложной задачей из-за отсутствия системного биомониторинга во многих странах, но данные токсикологических международных организаций позволяют составить примерное представление о проблеме. Например, по данным ВОЗ, в 2021г последствия воздействия свинца, в основном в виде ССЗ, стали причиной >1,5 млн. случаев смерти во всем мире. Установленной нормы поступления свинца в организм без вредных последствий для здоровья не существует. Свинец оказывает комплексное негативное влияние на миокард предсердий, воздействуя на ключевые механизмы клеточной регуляции: гомеостаз кальция, оксидативный баланс и вегетативную регуляцию. Это создает «идеальные» условия для развития ФП и других предсердных аритмий как за счет триггерной активности, так и за счет структурного ремоделирования. Учитывая повсеместное загрязнение и отсутствие безопасной концентрации, хроническая интоксикация свинцом должна рассматриваться как потенциально значимый фактор риска в кардиологической практике, особенно у пациентов с идиопатическими аритмиями [38].
Тяжелые металлы конкурируют с эссенциальными катионами. Так, свинец (Pb²⁺) конкурентно замещает кальций (Ca²⁺) в митохондриях, нарушая кальциевый сигналинг и индуцируя митохондриальную проницаемость; кадмий (Cd²⁺) замещает цинк (Zn²⁺) в антиоксидантных ферментах (супероксиддисмутаза), подавляя защиту от АФК. Кроме того, ионы тяжелых металлов напрямую ингибируют ферменты дыхательной цепи и антиоксидантной защиты: мышьяк (As) ковалентно связывается с тиоловыми группами (-SH) белков, инактивируя пируватдегидрогеназу и α-кетоглутаратдегидрогеназу (цикл Кребса), а также глутатионпероксидазу; Ртуть (Hg²⁺) необратимо ингибирует селенсодержащие ферменты, включая тиоредоксинредуктазу и глутатионпероксидазу. Некоторые металлы (свинец, алюминий) нарушают включение железа в гем, способствуя вторичному функциональному железодефициту внутри митохондрии. Все тяжелые металлы, попадая в организм, прямо или опосредованно генерируют АФК (через реакцию Фентона, ингибирование антиоксидантов), повреждая митохондриальные мембраны и ДНК, нарушая цепи митохондриального дыхания и синтеза АТФ. В общем, все тяжелые металлы действуют как мощные прооксиданты и метаболические яды, дестабилизирующие все ключевые митохондриальные процессы [39].
Распространенность хронической интоксикации тяжелыми металлами в популяции кардиологических, в т.ч. аритмологических пациентов, сильно недооценена профессиональным сообществом. А между тем именно ей могут быть объяснены случаи упорного течения ФП, неэффективности антиаритмической терапии и рецидивов после интервенционного лечения.
4. Хронический дефицит холекальциферола (витамин Д3)
Холекальциферол традиционно называется витамином Д, но с биохимической и функциональной точки зрения является типичным стероидным гормоном. Как и все стероидные гормоны (кортизол, альдостерон, половые гормоны), витамин Д3 синтезируется из холестерина. Активная форма (кальцитриол) действует через внутриклеточные рецепторы, регулируя экспрессию генов. Организм может полностью синтезировать витамин Д3 из 7-дегидрохолестерина в коже под действием ультрафиолетового излучения. Его активность регулируется сложной системой обратной связи, как у классических гормонов. Это понимание кардинально меняет подход к его роли в организме.
Давно известно, что витамин Д3 (холекальциферол) является ключевым регулятором системного кальций-фосфорного обмена. Однако его влияние на внутриклеточный гомеостаз кальция в КМЦ представляет собой отдельный малоизученный аспект, особенно важный для электрофизиологически уязвимых предсердий [40]. Гипотетически, дефицит витамина Д3, через прямые и опосредованные пути, может нарушать функционирование медленных кальциевых каналов КМЦ, создавая проаритмогенный фон. Длительный и клинически значимый дефицит витамина Д3 ведет к повышению уровня паратгормона, который мобилизует кальций из костей и увеличивает его реабсорбцию в почках, что усугубляет дефицит витамина Д и приводит к избытку фосфатов. Этот порочный круг создает нестабильный электролитный баланс в плазме крови, к которому чрезвычайно восприимчивы предсердные КМЦ [41].
Однако витамин Д3 давно перестал рассматриваться исключительно как регулятор кальций-фосфорного обмена. Современные исследования доказывают его роль как важного иммуномодулятора и регулятора воспалительных процессов. Рецепторы к витамину Д экспрессируются практически во всех иммунных клетках, что объясняет его широкое влияние на иммунный ответ.
Несколько рандомизированных клинических исследований у лиц с ожирением, предиабетом и метаболическим синдромом показали, что высокие дозы витамина Д (>4000 МЕ/сут.) могут улучшать чувствительность к инсулину, снижать уровень триглицеридов и подавлять синтез маркеров системного воспаления [41][42].
Кроме того, доказано, что витамин Д3 — физиологический ингибитор ренина. Его дефицит ведет к избыточной активности ренин-ангиотензин-альдостероновой системы, что повышает сосудистое сопротивление, снижает биодоступность эндотелиального оксида азота и индуцирует эндотелиальную дисфункцию.
В настоящее время есть данные клинических исследований, доказывающих прямую корреляцию между дефицитом витамина Д3 и встречаемостью ФП в общей популяции, а также позитивное влияние приема высоких доз витамина Д3 на профилактику ФП у пожилых. В ряде клинических исследований обнаружены его терапевтические эффекты на артериальную гипертензию и эндотелиальную дисфункцию, что, несомненно, может быть полезной дополнительной опцией в стратегии лечении пациентов с ФП [41-44].
5. Патологическая гиперсимпатикотония
Возникая как компенсаторный механизм при различных ССЗ, гиперсимпатикотония быстро трансформируется в самостоятельный патологический процесс, который через прямое кардиотоксическое, проаритмогенное действие, стимуляцию системного и органного воспаления ведет к усугублению ремоделирования миокарда. Нейромедиаторы СНС, в первую очередь норадреналин (норэпинефрин), напрямую, а также через активацию ренин-ангиотензиновой системы (ангиотензин II), стимулируют пролиферацию предсердных фибробластов и повышают синтез коллагена I и III типа, являясь, таким образом, индукторами диффузного интерстициального фиброза предсердий. Хроническая адренергическая стимуляция активирует пути, ведущие к гипертрофии предсердных КМЦ, и одновременно запускает митохондриальный кальций-опосредованный апоптоз, что приводит к потере жизнеспособных клеток и замещению их фиброзной тканью. Хроническая длительная гиперстимуляция β1-адренорецепторов миокарда приводит к тахикардии и усилению сократимости, резко увеличивая потребление кислорода миокардом предсердий. Одновременно симпатически опосредованная вазоконстрикция затрагивает и мелкие артерии предсердий, особенно в условиях эндотелиальной дисфункции. Патологическая гиперсимпатикотония индуцирует низкоуровневое системное воспаление, стимулируя выброс провоспалительных цитокинов (ИЛ-6, ФНО-α), активирует НАДФН-оксидазу, генерируя АФК. В итоге воспаление и оксидативный стресс повреждают клетки, усугубляют фиброз и дисфункцию ионных каналов, замыкая порочный круг ремоделирования предсердий [17][18].
Всё вышеперечисленное делает стратегии, направленные на снижение симпатического тонуса, патогенетически обоснованным и обязательным компонентом лечения и профилактики ФП, особенно у пациентов с СН, артериальной гипертензией и другими состояниями, сопровождающимися нейрогуморальной гиперактивацией. Блокада этого пути не только контролирует аритмию, но и замедляет прогрессирование основного заболевания.
Медикаментозная модуляция СНС путем назначения β-АБ не удовлетворяет всем требованиям, поскольку обладает рядом общеизвестных выраженных побочных эффектов и обширным перечнем противопоказаний. Кроме того, известно, что β-АБ имеют сродство к β-рецепторам клеток Ларгенганса, которые есть не только в поджелудочной железе, но и в коже, и в слизистой кишечника. Лечение β-АБ связано с индукцией или усугублением псориазоподобного воспаления кожи. Лизосомотропные β-АБ идентифицированы как критические индукторы ИЛ-23A в клетках Лангерганса, полученных из моноцитов человека, в условиях асептического воспаления. Доказано, что прием β-АБ специфически индуцирует продукцию АФК, критически важную для секреции ИЛ-23А в дермальных клетках Лангерганса, что ведет к значимому увеличению напряженности системного воспалительного ответа [45].
Кроме того, в 2025г были обнародованы результаты исследования REDUCE-AMI, которое заключалось в наблюдении >8500 пациентов после перенесенного инфаркта миокарда с сохранной фракцией выброса, подвергшихся реваскуляризации на протяжении 3,7 лет. Исследование показало, что длительное применение β-АБ не приводит к уменьшению рисков внезапной сердечной смерти и повторных инфарктов миокарда, а у женщин прием β-АБ увеличивает риск внезапной сердечной смерти на 2,5% по сравнению с контрольной группой [46].
В последние годы активно развиваются инвазивные и неинвазивные методики нейромодуляции, представляющие собой новую парадигму междисциплинарного взаимодействия интервенционной кардиологии и нейрохирургии/неврологии, смещающую фокус внимания с подавления активности субстрата аритмии на целенаправленную работу с ключевыми элементами вегетативной нервной системы и коррекцию нейрогенного звена патогенеза ФП, желудочковых аритмий и СН [47]. Пока ни одна из нейромодуляционных методик, несмотря на патогенетически обоснование принципы применения в лечении пациентов с ФП, не получила статус доказанной клинической эффективности и не входит в текущие рекомендации по лечению ФП. Отдельные клинические исследования инвазивной и неинвазивной стимуляции вагусного нерва и активации барорефлекса демонстрируют снижение маркеров системного воспаления у пациентов с ХСН, что гипотетически позволяет предположить возможность контроля над прогрессированием интерстициального и заместительного фиброза миокарда [47][48].
Стимуляция спинного мозга (ССМ), наряду с другими методиками также исследуется как потенциальный метод нейромодуляции для лечения ФП. Теоретическое преимуществом методики является способность ССМ активно воздействовать одновременно на оба звена эфферентной иннервации сердца, подавляя избыточную симпатическую активность, и одновременно активируя парасимпатическую, стимулируя вагусный нерв через афферентные пути, идущие от спинного к продолговатому мозгу. Прямое активирующее влияние ССМ на вагусный нерв с одновременным подавлением активности звездчатых ганглиев убедительно продемонстрировано в клинических исследованиях, посвященных лечению болевых синдромов [49]. У пациентов с ССМ статистически значимо снижалась плазменная концентрация норэпинефрина и адреналина и увеличивалась — ацетилхолина. Кроме того, в экспериментальных исследованиях выявлена способность ССМ контролировать реактивный глиоз задних рогов спинного мозга, возникающий в ответ на острую ишемию миокарда. Это приводит к подавлению нейровоспаления и системного воспалительного ответа, одновременно снижая экспрессию Fos-белков, являющихся маркером нейронального возбуждения и запускающих клеточный апоптоз. Регуляция экспрессии Fos-белков обеспечивает ограничение зоны постинфарктного фиброза [50]. На сегодняшний день большая часть убедительных данных получена в ходе доклинических исследований на животных моделях. Эти работы демонстрируют, что ССМ может снижать индуцибельность и продолжительность эпизодов ФП. Клинические данные использования ССМ при ФП у людей остаются крайне ограниченными; они в основном состоят из отчетов об отдельных случаях и небольших пилотных исследований [51-54].
Наш опыт применения ССМ у пожилых коморбидных пациентов с неврологической патологией (стеноз спинномозгового канала) и ФП составляет к сегодняшнему дню 23 человека, которые были прооперированы в нашей клинике в течение последних 9 лет (средний период наблюдение 3,6 лет, средний возраст на момент проведения операции имплантации нейростимулятора — 82,4 года). Критериями отбора были наличие клинически значимого стеноза спинномозгового канала в сочетании с ХСН 2-3 функционального класса по NYHA и персистентной формой ФП, резистентной к традиционному медикаментозному и интервенционному лечению. У всех наших пациентов мы стремились к тому, чтобы нейростимулятор работал не менее 12 ч в сутки на персонально подобранных комфортных параметрах стимуляции. Спонтанное восстановление синусового ритма мы получили у 15 пациентов в течение первых 12 мес. ССМ и еще у 3 пациентов — через 24 мес. постоянной стимуляции, что суммарно составило 78% эффективности. Кроме того, мы отмечаем постепенно снижение уровня промозгового натрийуретического пептида и уменьшение функционального класса ХСН в течение всего периода наблюдения у 21 из 23 пациентов, в т.ч. у 3 пациентов с хронической ФП. Механизм регресса ХСН, продолжающийся длительное время на фоне как синусового ритма, так и хронической ФП у пожилых пациентов не ясен.
Есть клинические и экспериментальные данные о том, что электрическое поле, возникающее при постоянной ССМ, является индуктором дифференцировки эндогенных стволовых клеток спинного мозга, и приводит к регенерации нервной ткани и восстановлению проведения после травматических разрывов спинного мозга [55][56]. Наша гипотеза заключается в том, что электромагнитное поле стимулирует дифференцировку стволовых клеток сердца, о существовании которых в миокарде известно [57]. На фоне ССМ на уровне С6-Th4, сердце попадает в зону воздействия электромагнитного поля, генерируемого на электродах, что, по аналогии с процессами, описанными в нервной ткани, может приводить к индукции дифференцировки стволовых клеток миокарда и, при условии длительного воздействия, обеспечивать частичный регресс интерстициального фиброза, приводя к улучшению параметров сердечной сократимости и снижению функционального класса ХСН и её лабораторных маркеров. К сожалению, большинство имплантированных нейростимуляторов у наших пациентов являются МРТ-несовместимыми, поэтому мы не можем визуализировать динамику состояния миокарда.
Заключение
На основе всего вышеизложенного, мы предлагаем дополнить стандартное обследование пациентов с ФП следующими пунктами:
- Маркеры низкоуровневого системного воспаления: С-реактивный белок ультрачувствительный, ИЛ-1β, -6, -23, ФНО-α.
- Маркеры латентного (аутоиммунного) миокардита: иммуноморфологическое исследование (титр антител к ядрам КМЦ, проводящей системе сердца, эндотелию сосудов); определение полимеразной цепной реакцией, IgM и IgG кардиотропных вирусов; МРТ миокарда с контрастным веществом в динамике.
- Маркеры латентного железодефицита: ферритин, растворимые рецепторы трансферрина, сывороточное железо (вне зависимости от уровня гемоглобина и эритроцитов).
- Развернутый анализ крови на гормоны щитовидной железы: Т3 общий, Т3 свободный; Т4 общий, Т4 свободный; ТТГ, антитела к тиреоглобулину и тиреопероксидазе.
- Анализ крови на витамин Д3 (25-ОН) и паратгормон.
- Токсикологическое исследование при упорном течении ФП, резистентной к стандартной терапии.
- Как обсуждаемая опция в комплексном обследовании — маркеры фиброза миокарда: лабораторное определение С-концевого пептида проколлагена I, N-концевого пропептида коллагена III, тканевого ингибитора матриксной металлопротеиназы (удобно для контроля терапии при условии их исходного повышения).
- При наличии технической возможности — исследование деформации миокарда предсердий (strain-эхо и strain-МРТ).
Выявление неочевидных факторов прогрессирования фиброза в миокарде предсердий позволит усовершенствовать стратегию персонализированного лечения пациентов с ФП и улучшить перспективу долгосрочной эффективности гайдлайн-обоснованной терапии.
Отношения и деятельность: автор заявляет об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье.
1. WHO guideline on use of ferritin concentrations to assess iron status in individuals and populations, 2020.
2. https://www.who.int/data/nutrition/nlis/info/iodine-deficiency.
3. Michael B. Zimmermann. Iodine and the iodine deficiency disorders. Present Knowledge in Nutrition (Eleventh Edition), Vol 1: Basic Nutrition and Metabolism, 2020, Pages 429-441.

Чтобы читать статью войдите с логином и паролем от scardio.ru
Ключевые слова
Для цитирования
Яковлева М.В. Нестандартные подходы к лечению фибрилляции предсердий — взгляд за рамки клинических рекомендаций. Мнение по проблеме. Российский кардиологический журнал. 2025;30(4S):6764. https://doi.org/10.15829/1560-4071-2025-6764. EDN: TIGAED
Скопировать