Генетические полиморфизмы, ассоциированные с развитием аритмического типа сердечно-сосудистых событий
Аннотация
В обзоре проанализирована глобальная повестка в отношении особенностей мутационного статуса генов, ассоциированных с развитием неблагоприятных сердечно-сосудистых событий аритмического типа, путем изучения подобранных данных из достоверных литературных медицинских источников. Полноэкзомное секвенирование генов позволит выделить группу риска по вероятности наступления ранних или отсроченных сердечно-сосудистых событий аритмического типа, в особенности среди пациентов, получающих терапию кардиотоксическими противоопухолевыми препаратами. Выделенная актуальная панель генетических полиморфизмов даст возможность оптимизировать подход к ведению пациентов, основываясь не только на клинических, лабораторно-инструментальных и анамнестических данных.
В настоящее время лидирующую позицию по причинам смертности среди населения занимает патология сердечно-сосудистой системы. По данным А. Д. Каприна, В. В. Старинского, А. О. Шахзадовой о «Состоянии онкологической помощи населению России в 2021 году», смертность от неонкологических причин среди пациентов со злокачественной нозологией составила 34,8%, что является весомым в масштабах страны. Чаще всего неблагоприятные события со стороны сердца среди данной группы пациентов связаны с терапией кардиотоксическими препаратами [1]. Несмотря на успехи в отношении выживаемости пациентов со злокачественными новообразованиями, в т.ч. онкогематологическими заболеваниями, препараты противоопухолевой терапии продолжают оказывать токсические эффекты на сердце, повышая смертность в безрецидивный период [2]. На сегодняшний день к нежелательным явлениям со стороны сердца после проведения курса противоопухолевой терапии относят систоло-диастолическую дисфункцию миокарда с динамически прослеживающимся снижением фракции выброса левого желудочка, разнообразные варианты аритмий, воспалительные заболевания сердца, артериальную и/или изолированную легочную гипертензию, ранние и/или отсроченные тромботические осложнения [3]. Одним из наиболее часто встречающихся первичных клинических проявлений дисфункции миокарда считают различные варианты аритмий [4]. К опасным состояниям для пациентов, находящихся в процессе терапии противоопухолевыми препаратами, следует отнести удлинение интервала QT, расстройство электрической активности сердца [5].
В условиях реальной клинической практики наиболее используемыми методами в отношении диагностики наступающей дисфункции миокарда являются как лабораторные: мониторинг маркеров ишемии миокарда (тропонины I, T, креатининфосфокиназа), анализ наступления и прогрессии хронической сердечной недостаточности (натрийуретический пептид), воспалительные изменения (миоглобин, аланинаминотрансфераза, аспартатаминотрансфераза, креатининфосфокиназа); так и инструментальные данные: оценка состояния объема миокарда, сосудов и окружающих тканей, клапанного аппарата, сократительной способности (электрокардиограмма, эхокардиографическое исследование, спеклтрекинговая эхокардиография, магнитно-резонансная томография сердца) [6]. Существующие способы диагностики изменений миокарда, в особенности у пациентов в процессе терапии противоопухолевыми кардиотоксическими препаратами, считаются недостаточными, выявляя нарушения со стороны сердца на этапе уже случившихся преобразований [7]. Однако в настоящее время методы диагностики продолжают совершенствоваться. Новым направлением в кардионкологии, открывающим возможности для специалистов смежных специальностей, считают изучение генетических полиморфизмов [8]. В зависимости от претерпевающих изменений в результате контакта организма с химиотерапевтическими агентами, ожидаются несколько разнообразных форм миопатий [9]. Согласно этому, генетические полиморфизмы, ассоциированные с развитием сердечно-сосудистых нежелательных явлений, можно распределить по фенотипам в зависимости от изменений, которые претерпевает миокард: сосудистому, гипертоническому, аритмическому, воспалительному. Деление генетической карты на категории позволит оптимизировать процесс диагностики ожидаемых нежелательных явлений со стороны сердца, в особенности на старте терапии онкологического заболевания.
Актуальность исследования в данном направлении определена на основании изучения генов, влияющих на метаболическую активность химиотерапевтического агента, биологию клеток миокарда, сосудов, микроокружение, вызывая нежелательные сердечно-сосудистые явления [10]. Существует ряд работ, в которых был проведен анализ небольшого количества полиморфизмов, достоверно ассоциированных с развитием кардиотоксичности, у пациентов, получающих терапию, в схему которой входили антрациклиновые соединения. Случаи антрациклининдуцированной кардиотоксичности, сочетанной с набором определенных полиморфизмов в генах АВСС2, HIF1A, CDKN2A/В и др., дает возможность предположить, что не только наличие общепринятых факторов влияет на вероятность возникновения кардиотоксических осложнений [11]. В настоящее время уже описана подгруппа мутационных изменений генов, которые участвуют в транспорте лекарственных препаратов и достоверно вызывают эндотелиои кардиотоксичность у пациентов, находящиеся на противоопухолевой терапии [12]. Новизна изучения предикторов кардиотоксичности основана на проведении полноэкзомного секвенирования, анализе и интерпретации генетических причин нарушений ритма сердца, что позволит полноценно сформировать индивидуальный подход к каждому больному и вырабатывать усовершенствованную тактику в отношении профилактики и превентивной диагностики сердечно-сосудистых событий.
Цель исследования: предоставить актуальные научные медицинские данные в отношении генетических полиморфизмов, ассоциированных с возникновением сердечно-сосудистых событий аритмического типа.
Материал и методы
Настоящий литературный обзор посвящен изучению актуальной научной медицинской информации в отношении генетических полиморфизмов, ассоциированных с развитием аритмического типа сердечно-сосудистых событий, в особенности, у пациентов онкогематологического профиля, получающих терапию противоопухолевыми кардиотоксическими препаратами, выявлению возможной области их применения и целесообразности введения в реальную клиническую практику. Поиск литературы по теме обзора осуществлен в базах данных Кокрановской библиотеки (https://www.cochranelibrary.com/), Medline (https://pubmed.ncbi.nlm.nih.gov/), научной электронной библиотеке (elibrary.ru).
На старте формирования литературного обзора проанализирован 101 литературный научный медицинский источник по запросу «генетические полиморфизмы в кардиологии». Критерии включения данных в список анализируемой литературы:
- Исследования, опубликованные на русском и английском языках, давностью не >5 лет (20182022);
- Высокая цитируемость медицинского литературного источника;
- Формат публикации исследования: клинические и экспериментальные исследования, оригинальные исследования, обзорные статьи, метаанализы.
Критериями исключения были: неполные исходные данные или исследования, в которых не отражена связь между генетическими полиморфизмами и изменениями в отношении миокарда; язык публикации, отличный от русского или английского языков; частный доступ к полнотекстовой информации из источника. Ключевыми словами на русском языке в строке поиска информации баз данных являлись: генетические полиморфизмы, аритмии, генетика в кардиологии; на английском — genetic polymorphisms, genetics in cardiology.
В результате селекции научно-медицинской информации, согласно предоставленным критериям, было отобрано 50 литературных источников.
Общий патогенез формирования аритмий
Аритмии — одна из самых частых нозологических форм патологии сердца, характеризующаяся разнообразием вариантов и вариабельностью осложнений.
Частой причиной возникновения аритмий является применение химиотерапевтических агентов, предназначенных для уничтожения опухолевых клеток посредством нарушения их метаболической и митотической активности [13]. В основе нежелательного явления терапии лежит прямое токсическое действие на кардиомиоциты, которое оказывается вследствие связывания противоопухолевых препаратов с сократительными белками сердца, кардиолипином, вызывая распад миофибрилл и мембранных молекул [14]. В результате вызванной «гибели» структур кардиомиоцита происходит нарушение баланса энергетического обмена клетки, адекватного транспорта ионов, необходимых для поддержания стабильного и регулярного сокращения сердечной ткани [15]. Наибольшее влияние на возникновение различных вариантов аритмий среди пациентов, получающих противоопухолевую терапию, оказывает неадекватная работа механизмов транспорта ионов. Одним из самых значимых факторов является сдвиг необходимых для нормального сокращения миокарда потока Са2+ (кальция), Na+ (натрия), К+ (калия) через мембрану клетки. Скопление молекул, ставшее фрагментированным, вызывает нарушение образования сердечного импульса, генерируя его сверхнормальное или недостаточное проведение, формирует основу для возникновения аритмогенного эффекта [16].
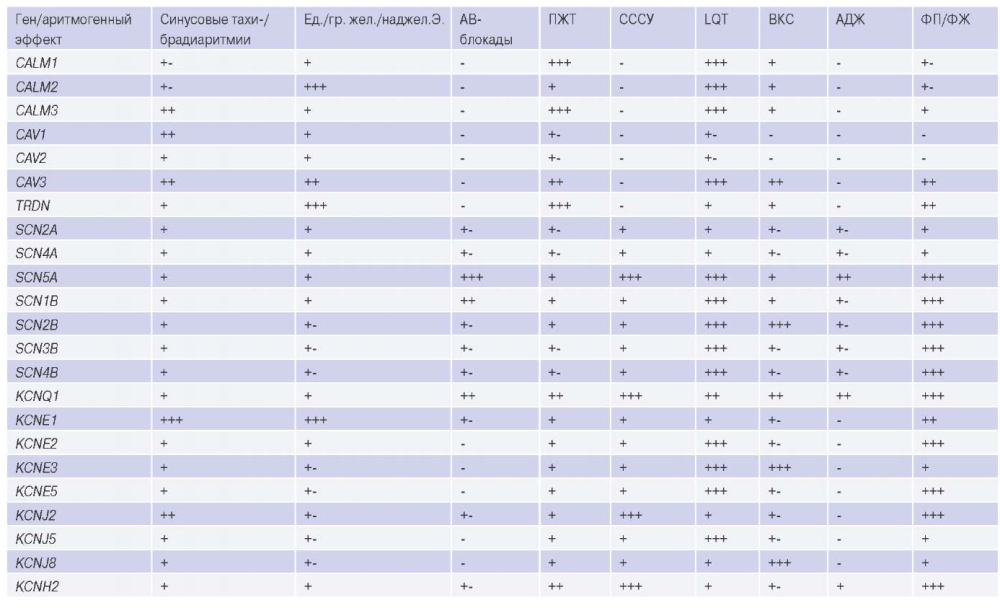
Генетические полиморфизмы, ассоциированные с развитием сердечнососудистых событий аритмического типа Учитывая несколько основополагающих механизмов, предрасполагающих к возникновению аритмий и разнообразные типы мутационного статуса, генетические полиморфизмы были сгруппированы по блокам, вызывающие идентичные изменения в работе микрои макрокомплексов миокарда. Наиболее распространенные виды аритмогенных эффектов, вызванных мутациями в том или ином гене, отражены в таблице 1.
Таблица 1
Виды аритмогенного эффекта в зависимости от полиморфизма исследуемого гена

Примечание: частота встречаемости: «+++» — очень часто, «++» — часто, «+» — редко, «+-» — спорный вопрос о частоте встречаемости, «-» — не встречается.
Сокращения: АВ-блокады — атриовентрикулярные блокады, АДЖ — аритмогенная дисплазия желудочков, ВКС — внезапная коронарная смерть, Ед./гр. жел./ наджел.Э. — единичные или групповые желудочковые или наджелудочковые экстрасистолы, ПЖТ — пароксизмальная желудочковая тахикардия, СССУ — синдром слабости синусового узла, ФП/ФЖ — фибрилляция предсердий или желудочков, LQT — синдром удлинённого QT.
Полиморфизм генов, предрасполагающих к кальмодулинопатиям
Нормальный генный продукт кальмодулин — это кальций-связывающий белок, способный ион-подчиняющим образом регулировать активность других белков. Его молекула состоит из двух глобулярных доменов, имеющих по два кальций-связывающих конца и специализированный центральный спиральный шарнир [17]. Несмотря на то, что кальмодулин, как единица, не проявляет ферментативной активности, белок является субъединицей множества ферментов, отвечающих за адекватность любой формы мышечной подвижности (протеинкиназа, протеинфосфатаза, фосфодиэстераза и др.) [18]. После связывания с ионами кальция кальмодулин приобретает высокое сродство к белкам-мишеням, присоединяется к ним и изменяет их каталитическую активность, влияя преимущественно на ионные каналы, аквапорины, а также множество других значимых для формирования электрического потенциала сердца белков [19].
Существует три отдельных гена CALM1, CALM2 и CALM3, каждый из которых кодирует идентичный полипептид из 149 аминокислот, мутации которых ассоциированы с риском развития неблагоприятных исходов со стороны сердца [20]. Идентифицировано несколько видов преобразований кальмодулина, большинство из них ассоциировано с измененной структурой в С-домене, в которой на первый план выступает замена высококонсервативных остатков аспарагиновой кислоты, необходимых для связывания ионов Ca2+. Такие преобразования в белке нарушают взаимодействие рецептора кальмодулина с рианодином (классом кальциевых каналов), вызывая дисфункцию потенциалов сердечного действия в различных клеточных моделях сердца [21]. Нарушения Ca2+-зависимой инактивации при дисфункции регуляции каналов L-типа вызывают стойкие изменения в электрофизиологии кардиомиоцитов человека. В совокупности неадекватной работы механизмов, волны иона кальция становятся фрагментированными, вызывая нарушение образования импульса, его сверхнормальное или замедленное проведение, обеспечивая аритмогенный эффект [22].
В результате полноэкзомого секвенирования насчитывают >50 видов нуклеотидных замен у CALM-положительных пациентов. Представленные генетические полиморфизмы могут вызывать жизнеугрожающие варианты нарушений ритма сердца. Кальмодулинопатии, ассоциированные с патогенными вариантами с участием трех генов кальмодулина CALM1, CALM2 и CALM3 проявляют себя значительным удлинением интервала QT, нарушают электрическую активность желудочков [23]. Основными клинико-инструментальными проявлениями таких изменений в структуре кардиомиоцитов можно считать атриовентрикулярные блокады, альтернации зубца Т, выраженные зубцы U, синусовые такии брадиаритмии, разнообразные варианты экстрасистол, желудочковую тахикардию, фибрилляцию предсердий и желудочков, внезапную коронарную смерть [24].
Полиморфизм генов, предрасполагающих к кавеолинопатиям
Кавеолы — небольшие образования в виде выпячивания плазматической мембраны большинства видов клеток, состоящие из ключевого белка — кавеолина и липидов разнообразных структур [25]. Кавеолины участвуют в ряде значимых для организма процессов, таких как эндоцитоз, метаболизм липидов, механосенсорная реакция и реакция выживания на стрессовые стимулы [26].
На сегодняшний день идентифицировано три основные изоформы кавеолинов и считается, что ген CAV3, в большей степени экспрессируясь в клетках сердечной мускулатуры, играет основную роль в процессах регуляции миокардиальной активности [27]. Генетические полиморфизмы, ассоциированные с мутациями в генах CAV1, CAV2, CAV3, которые выступают в качестве каркаса для организации молекул белков ионных каналов, нарушают не только передачу клеточных сигналов, но и создают условия для дисбаланса клеточной структуры [28].
Кавеолин-3 опосредует транспорт положительно заряженных ионов натрия, а кавеолин-1 взаимодействует с субъединицей калиевого канала Kir2.1. В совокупности данные процессы генерируют электрические сигналы с последующей их передачей, что обеспечивает постоянную поддержку нормального сердечного ритма [29]. К тому же гены CAV2 и CAV3 регулируют уровень атомов кальция в кардиомиоцитах, обеспечивая контроль адекватного сокращения и расслабления сердечной мышцы [30]. Нарушенный транспорт частиц и аномальная работа ионных каналов может изменить постоянство и регулярность электрической активности миокарда, приводя к нарушению сердечного ритма, характерному для синдрома удлиненного интервала QT [31].
Полиморфизм генов, вызывающих триадининопатии
Триадин — трансмембранный белок саркоплазматического ретикулума, в котором сосредоточен основной пул ионов кальция, экспрессирующийся в скелетных и сердечных мыщцах. Основной задачей данного белка является регулирование внутриклеточного потока ионов Ca2+. Триадин, кальсеквестрин и рианодиновый рецептор и образуют молекулярный комплекс, расположенный между саркоплазматическим ретикулумом и Т-трубочками [32]. Регуляция и обеспечение достаточным для адекватного сердечного сокращения количеством ионов кальция осуществляется путем влияния на связь кальсеквестрина и рианодиновых рецепторов [33]. При физиологическом уровне Ca2+, кальсеквестрин и рианодиновые рецепторы образуют прочную связь, которая препятствует открытию последних. При избытке ионов в саркоплазматическом ретикулуме, сцепление между макромолекулярным комплексом становится ослабленным, что приводит к открытию рианодиновых рецепторов и внутриклеточному высвобождению кальция [34]. Данные процессы играют ключевую роль в генерации возбуждения, адекватного сокращения и регуляции частоты сердечных сокращений.
Ген TRDN, который кодирует белок триадин, расположен на хромосоме 6q22-6q23, содержит 41 экзон и охватывает 420 тыс. пар нуклеотидов. Полиморфизмы данного гена (к примеру, делеция нуклеотидов в позиции 53ACAG или замена аминокислоты Thr59Arg) приводят к дефектам трансмембранного домена белка: его несостоятельности, утрате основных функций, вызывая так называемые триадининопатии [35].
Заболевания, связанные с полиморфизмом в гене TRDN, включают инверсию зубца Т, синдром сердечной аритмии, со слабостью скелетных мышц или без нее, катехоламинергическую полиморфную желудочковую тахикардию, синдром удлинённого интервала QT, внезапную коронарную смерть [36].
Полиморфизм генов, ассоциированных с развитием дисфункции натриевых каналов (sodium voltage-gated channel dysfunction)
Ионные каналы представляют собой белковые комплексы, осуществляющие перенос соответствующего иона согласно градиенту концентрации. Потенциал-зависимые Na+-каналы сердечной ткани состоят из связанного комплекса одной ?- и двух ?-субъединиц с разной молекулярной массой, где альфа-субъединица обеспечивает акти вность канала, а бета-субъединица модулирует кинетику инактивации канала [37]. Некоторые известные гены выборочно кодируют альфа (ген SCN2A, SCN4A и SCN5A) или бетта (гены SCN1B, SCN2B, SCN3B, SCN4B) субъединицу ионного канала миокарда. Комплексная работа единиц каналов обеспечивает проведение сокращения, координацию и поддержание нормального сердечного ритма [38].
Регуляторная ?-субъединица потенциалзависимых натриевых каналов играет важную роль в генерации возбуждения в мембранах головного мозга, сердца и скелетных мышцах. ?-субъединицы усиливают действие сестринской субъединицы на клеточной поверхности и корректируют состояние и скорость его инактивации [39]. Одни из самых распространенных полиморфизмов в генах SCN1B, SCN2B, SCN3B, SCN4B, кодирующих ?-субъединицы, зарегистрированы миссенс-мутации Val162Gly, Ile166Leu и Leu179Phe, которые связаны с удлинением продолжительности интервала QT на электрокардиограмме и высоким риском развития желудочковой тахикардии [40].
Наиболее клинически значимыми полиморфизмами считаются изменения в генах, кодирующих ?-субъединицу. Ген SCN5A, находясь на хромосоме 3 в регионе 3р22.2, состоящий из 28 экзонов, активно экспрессируясь в ткани миокарда, обеспечивает регуляцию альфа-субъединицы 5 типа и занимает лидирующую позицию в отношении возникновения аритмогенного эффекта в случае хромосомных перестроек. Распространенными миссенс-мутациями в заявленном гене считаются DelK1500, E1784K, Q779X, Т220I, R814W, D1595H [41]. Измененные структуры гена SCN5A способны вызвать два состояния в натриевом канале: состояние усиления потенциала действия или состояние утраты функции. Данные полиморфизмы вызывают некоторые жизнеугрожающие формы аритмий: синдром слабости синусового узла, синдром удлиненного интервала QT 3 типа, синдром Бругада, идиопатическую фибрилляцию желудочков, дилатационную кардиомиопатию и, как следствие, сердечную недостаточность [42].
Полиморфизм генов, ассоциированных с развитием дисфункции калиевых каналов (potassium voltage-gated channel dysfunction)
Существует несколько видов калиевых каналов, каждый из которых выполняет определенную функцию для обеспечения постоянства заданных процессов макроструктур: 1) Ca2+-активируемые каналы начинают свою работу в ответ на присутствие соответствующих ионов; 2) каналы внутреннего выпрямления транспортируют ионы калия внутрь клетки; 3) двухпоровый канал находится в непрерывно открытом состоянии и диктуют отрицательный мембранный потенциал; 4) потенциал-зависимые каналы, которые запускаются в ответ на изменение потенциала действия [43]. Потенциалзависимые калиевые каналы представляют более сложный класс ионных каналов, ориентируясь на структурную и функциональную стороны. Основная их задача состоит в регуляции процессов высвобождения нейромедиаторов, возбудимости нейронов, эпителиального транспорта электролитов; генерации и контроля частоты сердечных сокращений, обеспечение согласованной работы гладких мышц [44]. Белок KCNQ1, являясь членом подсемейства Q-зависимых каналов, взаимодействует с белками семейства KCNE (KCNE1, KCNE2, KCNE3, KCNE5) [45]. Четыре ?-субъединицы последних образуют структуру функциональных калиевых каналов, параллельно регулируя его активность. Наряду с крайними, белки KCNH2 и семейства KCNJ (KCNJ2, KCNJ5, KCNJ8) транспортируют положительно заряженные атомы К+ в клетки и из них [46]. Гены, кодирующие данные белки, принадлежит к вариабельному семейству генов, которые обеспечивают регуляцию создания адекватно функционирующих калиевых каналов. Наиболее изученными полиморфизмами, ассоциированными с возникновением аритмогенного эффекта, на сегодняшний день считаются R243W в KCNQ1, F57W в KCNE1, делеция эндогенного CRE11 в KCNH2 [47][48]. В миокарде полиморфизм генов, кодирующих соответствующие белковые структуры, дестабилизирует процесс «перезарядки» сердечной мышцы после каждого электромеханического сокращения, что, в свою очередь, приводит к нарушению регулярного ритма. Такие изменения в генетической карте пациента, находящегося в процессе терапии противоопухолевыми препаратами, могут достаточно рано привести к синдрому удлинённого и/или короткого интервала QT, мерцательной аритмии, аритмогенной дисплазии правого желудочка, острой коронарной смерти [49][50].
Заключение
Представленные в данном литературном обзоре полиморфизмы генов кодируют белки, необходимые для нормальной работы сердца. Существующие наработки в отношении взаимосвязи мутационного статуса генов и наступления нежелательных сердечно-сосудистых явлений определяют перспективность дальнейших исследований данного направления.
Отношения и деятельность: все авторы заявляют об отсутствии потенциального конфликта интересов, требующего раскрытия в данной статье.

Чтобы читать статью войдите с логином и паролем от scardio.ru
Ключевые слова
Для цитирования
Гиматдинова Г.Р., Данилова О.Е., Давыдкин И.Л., Хайретдинов Р.К., Антипова А.В. Генетические полиморфизмы, ассоциированные с развитием аритмического типа сердечно-сосудистых событий. Российский кардиологический журнал. 2022;27(3S):5069. https://doi.org/10.15829/1560-4071-2022-5069
Скопировать